Originally published May 3, 2017 and last updated Sep 24, 2020 This post was contributed by guest blogger, Addgene Advisory Board member, and Institute Scientist at the Broad Institute, John Doench. CRISPR technology has made it easier than ever both to engineer specific DNA ...
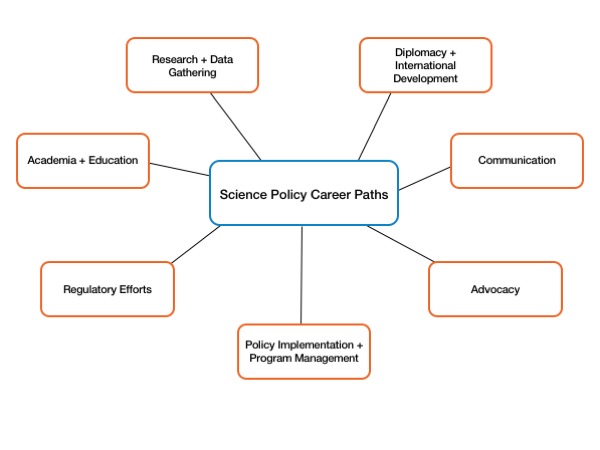
This post was contributed by Steph Guerra, a AAAS Science and Technology Policy Fellow at the Veterans Health Administration. “But, seriously, what even is science policy?” I have been asked this many times throughout my short science policy career and this seemingly simple ...
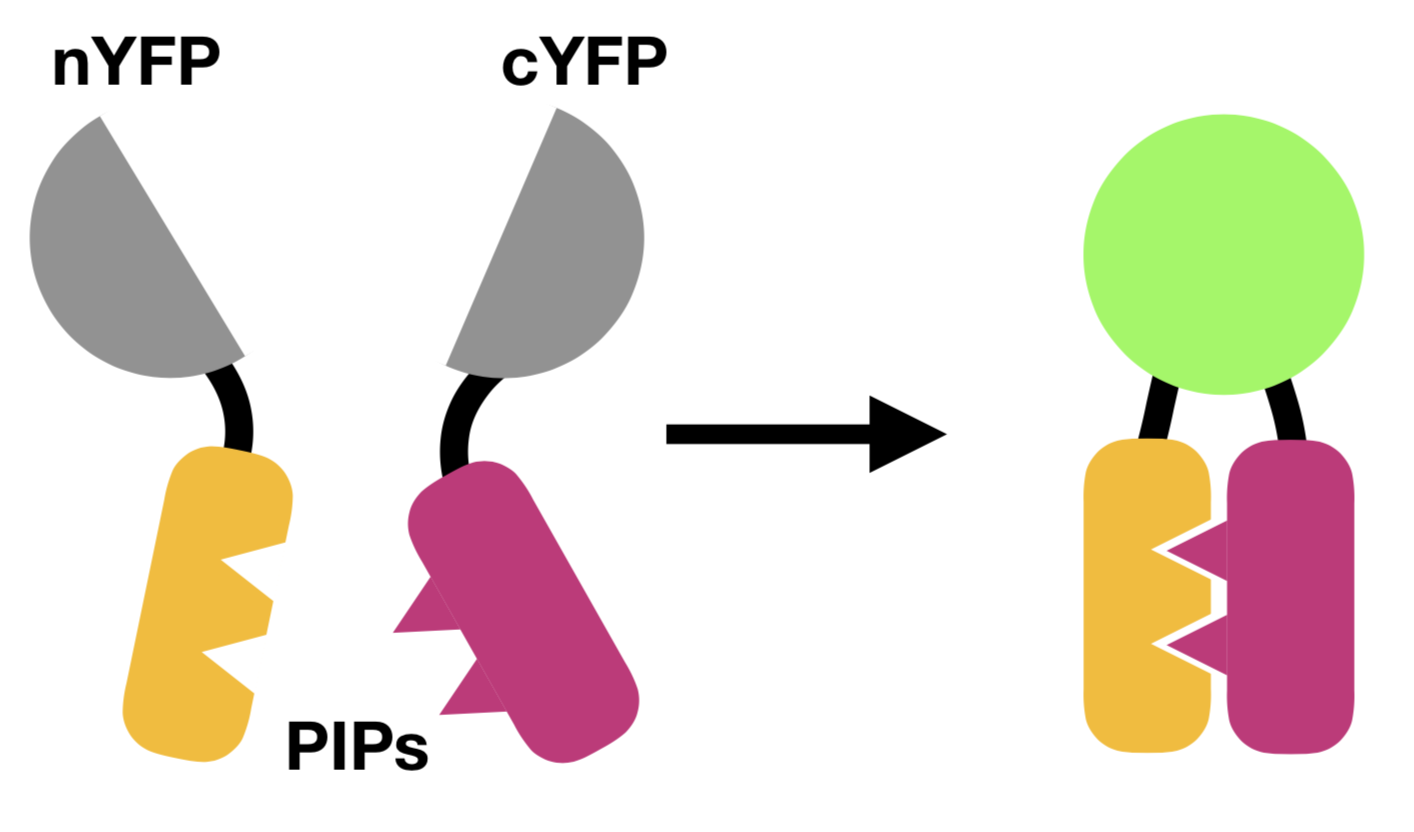
This post was contributed by Patrick Miller-Rhodes from the University of Rochester Medical Center. You’ve probably heard of Forster Resonance Energy Transfer (FRET). Through the non-radiative transfer for energy between neighboring fluorophores, FRET can be used to detect the ...
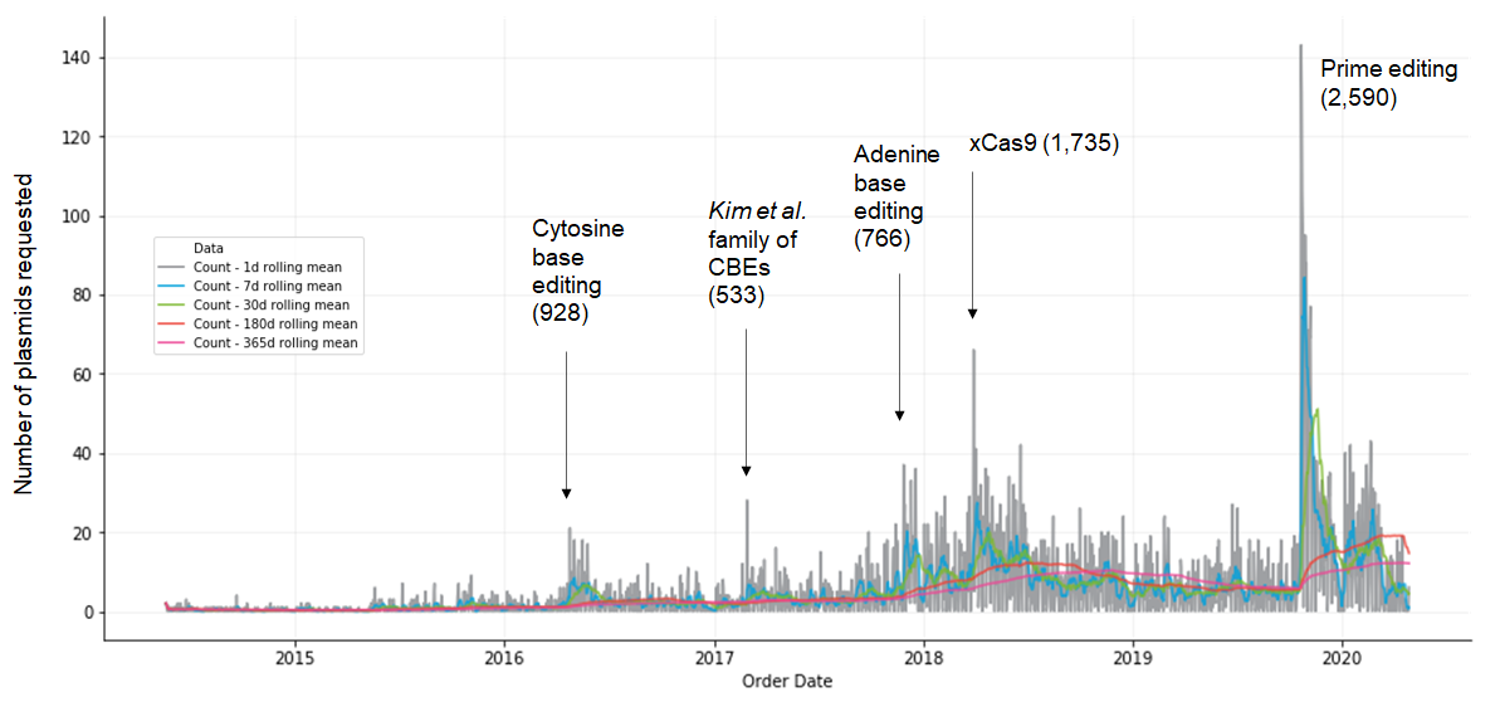
This post was contributed by Max W. Shen from MIT, Alvin Hsu Harvard University, and David R. Liu from the Broad Institute and Harvard University. Over the course of the last six months, COVID-19 has had a tremendous impact on our world -- as of June 4, 2020, COVID-19 has caused ...
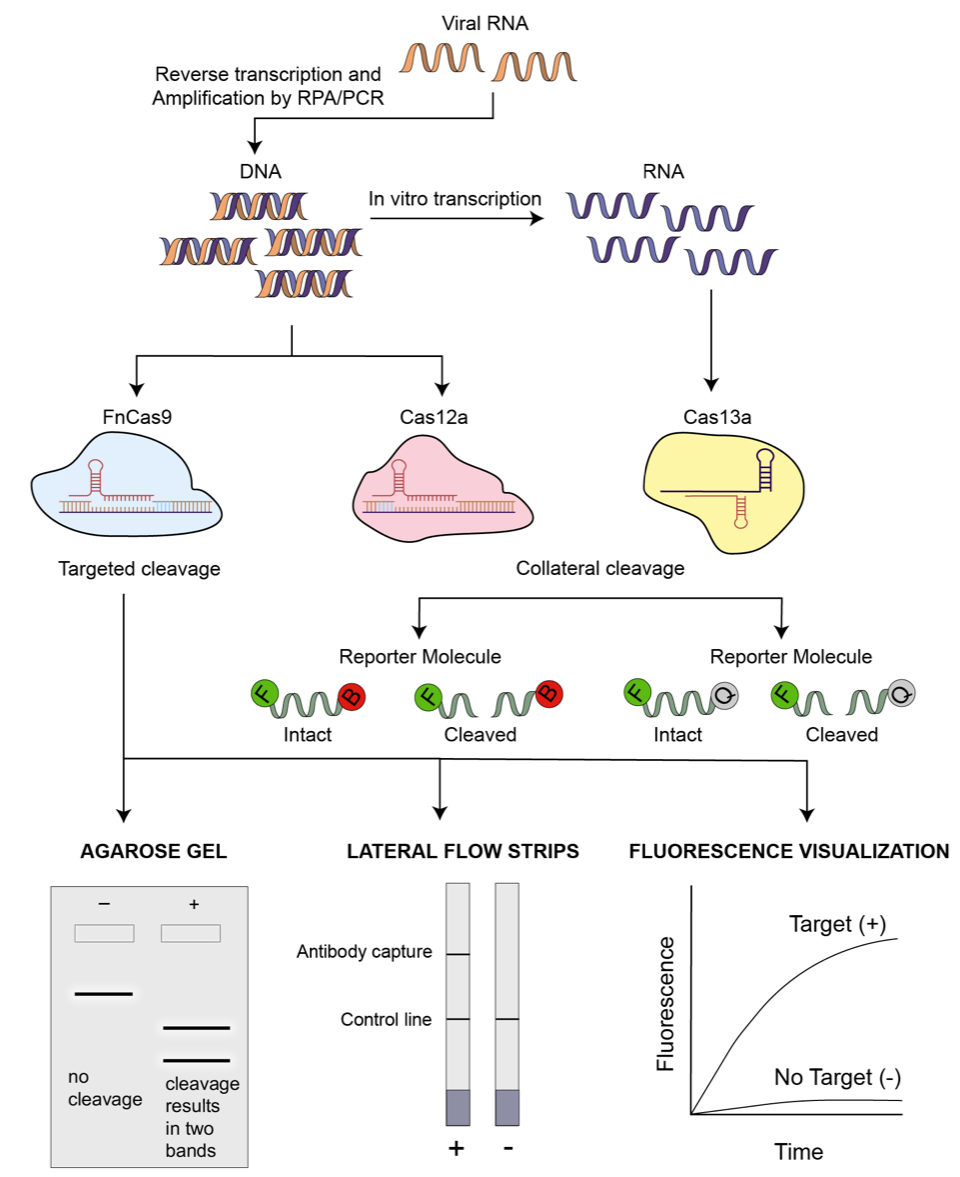
This post was contributed by Shravanti Suresh from Iowa State University. Since its appearance, SARS-CoV-2 has spread to almost every part of the world manifesting as a full-fledged pandemic. Containing the spread of this virus has become an utmost priority for countries around ...
This post was contributed by Manon Eckhardt and Melanie Brewer from the QBI Coronavirus Research Group at UCSF. It’s been only a few months since we all became acutely aware of the threat of SARS-CoV-2. Like many in the science community, we’ve been motivated to do anything and ...
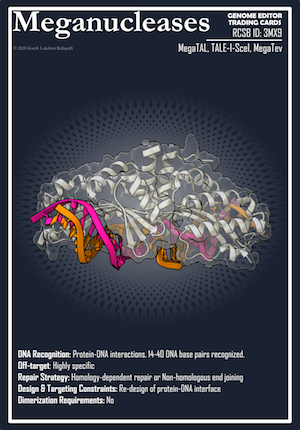
This post was contributed by Kartik Lakshmi Rallapalli, a graduate student at the University of California, San Diego. The revolution in genetic engineering techniques is a speculation of yesteryear which has been realized recently. Science Fiction (SciFi) writers have been ...
This post was contributed by Joe James from Binning Singletons. The sheer scale of a large conference can be intimidating. And it can be exacerbated when everyone seems to know one another, but they don’t know you. First time attendees and those attending alone often feel this ...