This post was contributed by guest blogger, Jessica Sacher, a microbiology PhD student at the University of Alberta studying with the Szymanski lab.
Reasons to study how a phage recognizes its host
Bacteriophages (viruses that prey on bacteria) may be the most numerous and most diverse biological entities on our planet, but we still know collectively little about how they infect and influence the evolution of their bacterial prey. Currently, receptor binding proteins (RBPs, the host recognition factors of phages) constitute one of the most popular classes of phage proteins to study. These are highly useful for the biotech industry, which is in the process of capitalizing on phage RBPs as diagnostic tools and therapeutics. In addition, the strategic use of whole phages as therapeutics, which is also gaining a lot of new traction lately (1, 2), depends on knowledge of the structure(s) a given phage will recognize on a host cell.
To make matters even more interesting, much of what a phage faces as it approaches a host cell surface are complex polysaccharide structures, many of which are easily varied by the cell and whose structures are just beginning to be understood. Therefore, in addition to their utility in the health sector, phages represent a reservoir of carbohydrate binding proteins waiting to be explored and exploited. With better characterization, these proteins could function as surrogate antibodies for identifying and characterizing sugars and sugar-containing molecules in molecular biology labs (for a review, see Simpson et al., 2015).
Visit Addgene's MIcrobiology Pages
The problem: Host binding proteins are hard to find
Sequencing and cloning phage DNA can be difficult, in part because phages frequently alter their DNA by adding methyl groups, glycosyl groups, and other modifications to their bases in order to protect their DNA from host cell nucleases (4). To further complicate matters, even when sequenced, phage genomes tend to be choc-full of open reading frames that display little to no homology to anything else, making it difficult to quickly identify possible gene function. In this post, I will describe a useful and cost-effective method that our lab (the Szymanski lab, which has recently moved from the University of Alberta in Edmonton, Canada, to the University of Georgia, USA) has developed that allows for a phage genome to be probed for proteins with host-binding activity without the need for genome sequencing or annotation (5). In the event that genome sequencing can be done, this technique can also help pinpoint the genes within a sequenced genome that encode for functional cell-binding proteins in the (all-too-common) event that homology-based searches return no candidates.
Our strategy: Express random fragments of the phage genome and probe with live cells
The following is a description of a technique published by Simpson, Sacher, and Szymanski in January 2016 in Viruses (5). Please refer to the paper for more details on the assay, including examples of RBPs successfully identified using this method.
Step 1: Prepare a random fragment library of phage DNA
This first step involves digesting phage genomic DNA into 1-3 kb (approximately gene-sized) fragments so that these fragments can be cloned into E. coli and subsequent proteins screened for cell-binding abilities (6).
This can be accomplished using a restriction enzyme with a short recognition sequence (like MluCI) that will cleave DNA frequently enough to result in a smear of fragments that get smaller in size as a function of digestion time. For phage DNA that is refractory to enzymatic digestion, however, a physical fragmentation strategy is preferable. Nebulization involves forcing DNA through a small opening using compressed nitrogen gas; this shears DNA at random locations and allows for a given fragment size to be selected for by manipulating the nebulization time and pressure. Nebulization units, which can be purchased cheaply, are small plastic containers that can easily be attached with a hose to a nitrogen gas tank with a pressure regulator. Other methods for physical shearing exist as well, such as sonication and needle shearing. Essentially, any method that results in the bulk of resultant fragments falling between 1 and 3 kb results in optimal likelihood of capturing a whole RBP gene without causing issues associated with cloning large fragments.
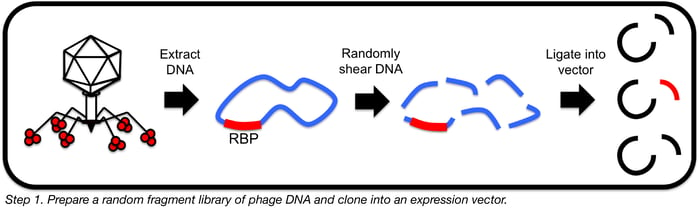 After fragmentation, repair sheared ends with T4 DNA polymerase (to create blunt ends) and ligate into a bluntly digested vector designed for protein overexpression in E. coli, such as pET30a. If working with DNA that is refractory to enzymatic manipulation, amplifying phage DNA with phi29 polymerase prior to fragmentation can greatly facilitate downstream manipulations. Phi29 polymerase is a phage-derived polymerase that can perform whole genome amplification (by a mechanism known as multiple displacement amplification) even in the presence of certain modifications to DNA bases.
After fragmentation, repair sheared ends with T4 DNA polymerase (to create blunt ends) and ligate into a bluntly digested vector designed for protein overexpression in E. coli, such as pET30a. If working with DNA that is refractory to enzymatic manipulation, amplifying phage DNA with phi29 polymerase prior to fragmentation can greatly facilitate downstream manipulations. Phi29 polymerase is a phage-derived polymerase that can perform whole genome amplification (by a mechanism known as multiple displacement amplification) even in the presence of certain modifications to DNA bases.
Ligate and transform your resultant plasmid libraries into chemically competent E. coli and analyze transformants for appropriately sized inserts. We consider a library worth using if at least 40% of colonies tested showed correctly sized inserts following colony PCR of 10-20 colonies using primers bordering the multiple cloning site of the vector. In order to reduce the chances of some clones being outcompeted by others in liquid culture, dilute and plate the complete library of transformants for single colonies (this usually requires 10-20 plates) (see box below). This initial transformation should be performed using a highly competent strain that you can use for long-term library storage. After overnight growth, scrape up, pool and extract plasmid DNA directly from the colonies. Transform the resultant plasmid DNA into E. coli BL21 (DE3) cells (these have the T7 polymerase and a few proteases knocked out making them particularly useful for protein production), then dilute and plate transformants for isolated colonies (150-200 per plate) across 20 fresh selective plates.

Step 2: Express random phage DNA fragments in E. coli
Now that random DNA fragments have been cloned into E. coli cells, isolated colonies will be lifted onto nitrocellulose membranes and exposed to IPTG in order to induce protein expression. Cells on the membranes will then be exposed to a lysis buffer to release recombinant proteins directly onto the nitrocellulose for subsequent probing for cell-binding activity.
First, cut out 20 Petri dish-sized circles of nitrocellulose membrane and press each gently down onto a plate of colonies. Mark the orientation of each membrane on its plate before moving on. Then peel each membrane off the plate (saving the plate so you can regrow the lifted colonies overnight), flip the membrane over and place it colony-side up onto a fresh LB plate containing an appropriate inducing agent (0.4 mM IPTG is a good start if you’re using a pET vector) and antibiotic for plasmid selection. Remove air bubbles between the membrane and the agar using forceps. Several protein induction conditions may need to be tested, as with any protein purification, but overnight at 30°C is a good start.
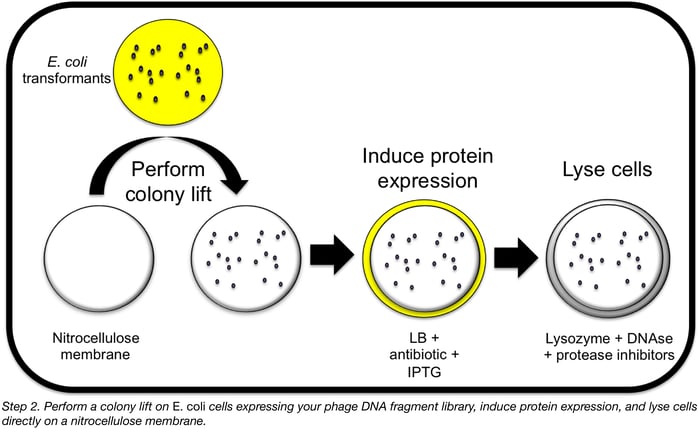
Prepare a lysis solution containing your favourite bacterial protein extraction reagent (BPER from Thermo works well) with some added lysozyme, DNAse I, and protease inhibitor cocktail. Line the bottom of 20 empty Petri dishes with thick filter paper and saturate each with lysis solution (~3 ml each), pouring off any excess.
When your protein induction is done, peel the membranes off the plates and place each onto a lysis solution-soaked filter paper (colony-side up) and leave for 1 h at room temperature. After this, it is helpful to subject your membrane to several freeze/thaw cycles to facilitate cell lysis: alternating between 5 min at -20°C (or -80°C) followed by 5 min at 37°C (each repeated 5 times) works well. Leaving the membrane overnight in the freezer during one of these steps is also a convenient stopping (ie. going home and eating and sleeping) point for this experiment.
Find Bacterial Expression Vectors in the FX Cloning System Kit
Step 3: Probe random phage DNA fragments with live host cells
Now you can proceed with incubating this membrane with your cells of interest and growing them up overnight. Assuming they expressed well, the proteins encoded by the plasmids in the library will remain attached to the membrane at the same location that their host colonies grew. If your cells of interest stick to the same location as a given protein, you have a potential receptor binding protein hit and can go back to the original plate to identify which phage gene encodes that protein!
First, block your membranes with BSA or skim milk as you would for a Western blot in order to reduce the number of cells that will bind non-specifically to either the membrane itself or to any “sticky” proteins expressed by your library. After blocking for 1 h at room temperature with gentle shaking, gently wick off excess liquid from each membrane and press a fresh Kimwipe down firmly onto each to remove cell debris. Then, UV-irradiate the membranes for 15 min to kill any remaining live E. coli. At this point, it can really help to incubate the membranes overnight in 500 mM to 1 M NaCl in order to enhance folding of the recombinant proteins. Optimal salt concentration will vary with each protein, but several of the phage RBPs we have worked with tended to bind cells more effectively in the presence of high salt.
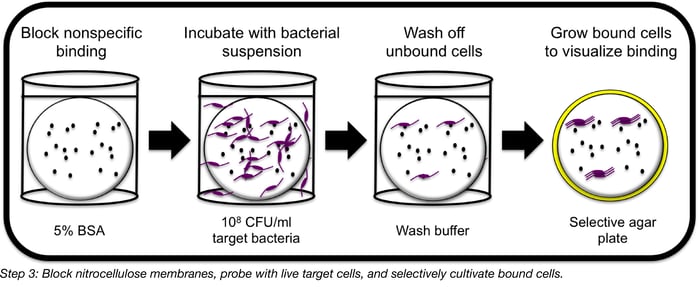
To probe membranes with target bacterial cells, wash your membranes with PBST to remove salt, then suspend a freshly harvested dose of cells (preferably in log phase) in blocking solution (around 108 CFU/mL is a good place to start) and add your membranes to the solution, shaking gently for 1 h at room temperature. This will allow any RBPs expressed on the membrane to bind their target cells, pulling them out of solution. Wash your membranes well with PBST to remove unbound cells.
To detect bound cells, simply lay each membrane on an agar plate selective for your target organism and incubate overnight according to appropriate cell growth conditions. Note: if using fast-growing bacteria like E. coli or Salmonella, or any cells that bind strongly to nitrocellulose, over-incubation of plates can lead to high background that obscures the desired signal. We have successfully corrected this by incubating plates for around 8-12 h at room temperature as opposed to overnight at optimal growth temperature. If you’re lucky enough to have an antibody or other protein on hand that will specifically bind your target organism, it can be helpful to spot this protein directly onto nitrocellulose and carry out the steps from blocking the membrane to probing with cells in order to optimize this portion of the assay before attempting it with a random library.
Following incubation, you should have visible colonies growing on the membrane wherever an E. coli colony expressing a functional binding protein was originally located. Now you can grow up your colonies of interest, sequence the inserted DNA and characterize your new putative host cell-binding proteins!
Many thanks to our guest blogger Jessica Sacher!
 Jessica Sacher is a microbiology PhD student at the University of Alberta studying with the Szymanski lab (though she’s tagged along with the lab during its recent move to the University of Georgia). She enjoys pondering how phages get through the day, spending time with her horse, and wondering what her cat is thinking. Find her on Twitter @JessicaSacher.
Jessica Sacher is a microbiology PhD student at the University of Alberta studying with the Szymanski lab (though she’s tagged along with the lab during its recent move to the University of Georgia). She enjoys pondering how phages get through the day, spending time with her horse, and wondering what her cat is thinking. Find her on Twitter @JessicaSacher.
References
1. Nobrega, Franklin L., et al. "Revisiting phage therapy: new applications for old resources." Trends in microbiology 23.4 (2015): 185-191. PubMed PMID: 25708933.
2. Cooper, Callum J., Mohammadali Khan Mirzaei, and Anders S. Nilsson. "Adapting drug approval pathways for bacteriophage-based therapeutics."Frontiers in Microbiology 7 (2016). PubMed PMID: 27536293. PubMed Central PMCID: PMC4971087.
3. Simpson, David J., Jessica C. Sacher, and Christine M. Szymanski. "Exploring the interactions between bacteriophage-encoded glycan binding proteins and carbohydrates." Current opinion in structural biology 34 (2015): 69-77. PubMed PMID: 26275959.
2. Weigele, Peter, and Elisabeth A. Raleigh. "Biosynthesis and Function of Modified Bases in Bacteria and Their Viruses." Chemical Reviews (2016). PubMed PMID: 27319741.
3. Simpson, David J., Jessica C. Sacher, and Christine M. Szymanski. "Development of an Assay for the Identification of Receptor Binding Proteins from Bacteriophages." Viruses 8.1 (2016): 17. PubMed PMID: 26761028. PubMed Central PMCID: PMC4728577.
4. Schmitz, Jonathan E., et al. "Rapid DNA library construction for functional genomic and metagenomic screening." Applied and environmental microbiology 74.5 (2008): 1649-1652. PubMed PMID: 18083885. PubMed Central PMCID: PMC2258639.
Resources on the Addgene Blog
- Learn How CRISPR Can Be Combined with Phage for Antimicrobial Purposes
- Use Yeast Two Hybrid Systems to Probe Protein-Protein Interactions
- Use CRISPR For Genome-Wide Screens
Resources on Addgene.org
- Visit Our Microbiology Pages
- Use the FX Cloning Sytem to Clone Your Gene of Interest Into a Variety of Expression Vectors
- Deposit Your Microbiology Plasmids

Topics: Microbiology, Molecular Biology Protocols and Tips, Plasmids





Leave a Comment