 This post was contributed by guest blogger, Kristian Laursen from Cornell University.
This post was contributed by guest blogger, Kristian Laursen from Cornell University.
Site directed mutagenesis is a highly versatile technique that can be used to introduce specific nucleotide substitutions (or deletions) in a tailored manner. The approach can be used in conventional cloning (to introduce or remove restriction sites), in mapping of regulatory elements (to mutate promoters/enhancers in reporter constructs), in functional analysis of proteins (to perform alanine scanning mutagenesis or targeted substitution of key residues), and in SNP analysis (to introduce naturally occuring SNPs in a plasmid context). The technique is also highly relevant in this age of CRISPR; site-directed mutagenesis generally applies to plasmids, but may also facilitate genome editing. Tailored mutations are commonly introduced to endogeneous DNA through homology-directed repair (HDR) of a CRISPR/Cas9 induced double-stranded break. This site-directed genome editing requires a template of high homology to the endogenous target, yet to facilitate the repair, the template should be resistant to Cas9 cleavage. If a plasmid contains the template, site-directed mutagenesis can be used to mutate the PAM sequence (an NGG sequence critical for Cas9 cleavage), thereby rendering the resulting construct resistant to Cas9 induced cleavage.
Summary of Site Directed Mutagenesis
In brief, point-mutations can be introduced to plasmids using primers (with the desired mutation) in a PCR protocol that amplifies the entire plasmid template. The parent template is removed using a methylation-dependent endonuclease (i.e. DpnI), and bacteria are transformed with the nuclease-resistant nicked plasmid (the PCR product). Plasmids are isolated from the resulting colonies, and screened for the desired modification. Finally, the positive clones are sequenced to confirm the desired modification and the absense of additional modifications.
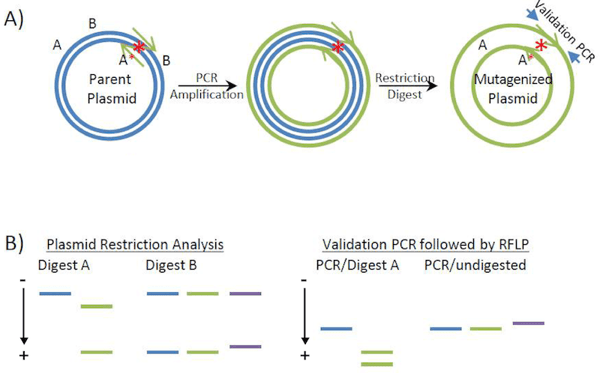 |
|
Site-directed mutagenesis of Plasmids. A) Generation of mutagenized plasmids. PCR primers (green) amplify the plasmid template (blue), and introduce a restriction site “A*” (in addition to the “A” site already in the parent vector). Also shown are the “B” restriction sites, which flank the targeted region. Upon PCR amplification, the template is removed by DpnI restriction digestion leaving behind only the mutagenized plasmid. Although it is commonly believed that the final products of the PCR reaction are pieces of double stranded, circular DNA with staggered nicks, recent data (see Xia et al., 2015) suggests that the final PCR products are pieces of linear, double stranded DNA with homologous ends. These homologous ends recombine in vivo to give the final circular plasmid product pictured here. B) Screening of recovered plasmids for the mutation of interest. Gel electrophoresis diagrams of the anticipated restriction analysis results (left) and validation PCR followed by Restriction Fragment Length Polymorphism (RFLP) analysis (right). Introduction of restriction site “A*” results in an additional band when site directed mutagenesis products (green) are digested with enzyme “A” as compared to the products of parental plasmid digestion (blue). Digestion with enzyme “B” detects primer multimerization (purple) which is evident by an increase in the size of the lower band (portion of the plasmid containing the mutation). Similar results can be obtained with PCR screening using the primers specified by the arrowheads in (A). |
Experimental guidelines
Primer Design
As a rule of thumb, 11 bp of complementary sequence on either side of the desired mutation (usually 1-3 mismatched bases) is sufficient for your primers to successfully anneal to the plasmid of interest during the PCR reaction. Ideally, your primers should be free of palindromic and repetitive sequences, but if present, a minor extension can usually ensure that the 3’-base(s) do not form secondary structures. The introduction (or ablation) of a restriction site through mutagenesis vastly facilitates the subsequent process of screening for succesfully mutated clones. Forward and reverse primers are designed to be complementary, but each primer may extend beyond the complementary region as long as an overlap with a minimum 6 bp is maintained. This overlap ensures that the PCR generates a nicked circle rather than a linear product (see figure).
Template
A high purity plasmid prep significantly increases the success rate of site directed mutagenesis. In addition, you may want to try different concentrations of template (e.g. 0.1-1.0 ng/μl). Smaller plasmids (~3 kb) are generally more efficiently amplified than larger constructs, but plasmids as large as ~6 kb can be amplified fairly easily by simply following the polymerase manufacturers’ PCR protocol. Be sure to adjust the extension time to match the size of your template. Amplification of GC-rich plasmids is facilitated by the addition of DMSO to the PCR reaction (usually around 3% final concentration). The DMSO reduces secondary structures of the DNA template, and may also decrease the annealing temp of the primers. Because you will be using a methylation-dependent enzyme (DpnI) to eliminate the parent plasmid from the PCR products, the plasmid template should be isolated from a methylation competent bacterial strain (e.g. DH5α which is dam+). Methylation deficient bacterial strains can be identified by the dam13(-) mutation – you’ll want to steer clear of these strains when preparing the plasmid template for site directed mutagenesis.
Polymerase
To ensure that you don’t introduce undesired mutations through the PCR process, you need a high-fidelity polymerase. There are many high fidelty polymerases on the market; you need one with 5’->3’ polymerase activity (for amplification), 3’->5’ exonuclease activity (increases amplification fidelity), and no 5’->3’ exonuclease activity (which could potentially truncate the template). Importantly, you also need a DNA polymerase that produces blunt-ended PCR products (e.g. Phusion, Pfu, and Vent polymerases). Note that certain polymerases such as Taq generate A-overhangs (a feature used in TA-cloning). Such non-complementary bases at the 3’-end interfere with plasmid reconstitution, and consequently Taq polymerases cannot be used for site directed mutagenesis.
Nuclease
To remove the template DNA (unmodified plasmid) a restriction digest with DpnI is used. DpnI is unique in that it cleaves only DNA that is methylated at the adenosine of the GATC recognition site.
Transformation: After the PCR reaction, no ligation is required since the E. coli you transform your PCR products into will efficiently patch up the DNA. The resistance marker from the parental plasmid provides a mean for selecting for transformants which have taken up the mutagenized plasmid. Note that any residual parental plasmid (usually from incomplete DpnI digest) can also form colonies under these conditions.
Screening
If a restriction site was introduced (or ablated), bacterial colonies can be screened by identifying the presence (or absence) of that particular site with fragment length polymorphism (RFLP) analysis. In this process, if your mutation introduces an additional restriction site into your plasmid, then, when the plasmid is digested with the appropriate restriction enzyme and run on a gel, one of the bands present in the digest products of the unmodified plasmid will be split into two smaller bands (Figure B, digest A). In contrast, if your mutation ablates a restriction site, digestion with the appropriate enzyme will merge two smaller bands visible in a digest of the unmodified plasmid into a larger band (not shown). As a footnote; a similar approach can be used to identify Cas9/CRISPR-induced genome modifications.
Occasionally, multimerization of the PCR primers can cause duplication of the primer sequence in the resulting plasmid. An additional restriction digest, which excises a short region (<400 bp) proximal to the target site, can identify these duplications (slightly larger bands sizes relative to the original template). Due to the small difference in size, the fragments should be separated on a high percentage agarose gel (~3%). Note that primer duplication will evade detection in the initial RFLP analysis simply because the introduced restriction site is also duplicated.
Validating your mutagenized plasmids
Once constructs containing the desired modification have been identified (and confirmed to be free of primer duplications), final validation is performed by sequencing functional regions of the plasmid including the insert. This is essential since mutations are potentially introduced anywhere in the plasmid (albeit at extremely low frequency), and these could interfere with the function of the resulting construct. In cases where the functionally important regions span several kb (or are GC-rich and therefore difficult to sequence), it may be easier to subsequently clone a smaller region (containing the specific modification) into the parental plasmid (or other). This is particularly relevant in cases where a large plasmid with multiple functional regions is used as template for the mutagenesis reaction.
Alternative approaches to site directed mutagenesis
Point-mutagenesis is fairly easy, but the risk of PCR-introduced mutations can make alternative approaches more favorable if you want to introduce a point mutation in a large construct. Particularly, if a pair of unique restriction site is located close to your target site, you can simply amplify and mutate this smaller portion of the plasmid with a primer pair that generates a linear product. The mutated product can then be cloned back into the original plasmid using the unique restriction sites. Alternatively, if you want to make your protein non-functional, you may simply introduce a frame-shift mutation. This can be achieved using a restriction enzyme that recognizes a unique site in the coding region (preferably near the 5’-end) and that also generates 4 bp overhangs. You can then use the Klenow fragment of DNA polymerase I to generate blunt ends from these overhangs and ligate these blunt ends together in a standard ligation reaction resulting in a frame-shifted coding region. It is worth noting that unlike these alternatives, site directed mutagenesis by PCR is not dependent on the proximity of unique sites and allows for the introduction of specifically designed mutations (not limited to frame shift mutations). Consequently, site directed mutagenesis is one of the most popular approaches for introducing minor modifications to a plasmid template.

Many thanks to our Guest Blogger Kristian Laursen!

Kristian Laursen is a researcher at Cornell University.
References
1. Breslauer, Kenneth J., et al. "Predicting DNA duplex stability from the base sequence." Proceedings of the National Academy of Sciences 83.11 (1986): 3746-3750. PubMed PMID: 3459152. PubMed Central PMCID: PMC323600.
2. Chester, Nicholas, and Daniel R. Marshak. "Dimethyl sulfoxide-mediated primer Tm reduction: a method for analyzing the role of renaturation temperature in the polymerase chain reaction." Analytical Biochemistry 209.2 (1993): 284-290. PubMed PMID: 8470801. PubMed PMID: 8470801.
3. Saiki, R. K., et al. "Primer-directed enzymatic amplification of DNA." Science 239 (1988): 487-491. PubMed PMID: 2448875.
4. Xia, Yongzhen, et al. "New insights into the QuikChangeTM process guide the use of Phusion DNA polymerase for site-directed mutagenesis." Nucleic acids research (2014): gku1189. PubMed PMID: 25399421. PubMed Central PMCID: PMC4333370.
Resources on the Addgene Blog
- Brush up on Your Cloning Techniques
- Learn More about Plasmids in Our Plasmids 101 Series
- Use CRISPR to Generate Specific Genomic Mutations
Resources on Addgene.org
Topics: Molecular Biology Protocols and Tips, PCR, Plasmids





Leave a Comment