Restriction enzyme cloning is the workhorse of molecular cloning; however, one of its biggest limitations is that sequence modifications can only be made at restriction enzyme cut sites. The lambda red system is an alternative method that can be used for cloning or genome engineering and is based on homologous recombination. It allows for direct modification of DNA within E. coli and is independent of restriction sites. The lambda red system is derived from the lambda red bacteriophage and its use as a genetic engineering tool is frequently called recombineering - short for homologous recombination-mediated genetic engineering. It can be used to make an assortment of modifications: insertion and deletion of selectable and non-selectable sequences, point mutations or other small base pair changes, and the addition of protein tags. It also has the flexibility to modify the E. coli chromosome, plasmid DNA or BAC DNA.
To use the lambda red recombineering system to modify your target DNA, you first electroporate a linear donor DNA substrate (either dsDNA or ssDNA – see below) into E. coli expressing the lambda red enzymes. These enzymes then catalyze the homologous recombination of the substrate with the target DNA sequence. This means cloning occurs in vivo, as compared to restriction enzyme cloning where the genetic changes occur in a test tube. The donor DNA substrate only requires ~50 nucleotides of homology to the target site for recombination.
Key components of the Lambda Red recombineering system
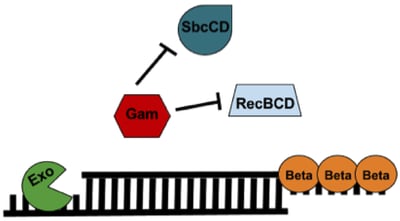
The lambda red recombineering system has three components (Figure 1): 1) Exo, 2) Beta, and 3) Gam. All three are required for recombineering with a dsDNA substrate; however, only Beta is required when generating a modification with an ssDNA substrate.
Gam: Gam prevents both the endogenous RecBCD and SbcCD nucleases from digesting linear DNA introduced into the E. coli.
Exo: Exo is a 5’→3’ dsDNA-dependent exonuclease. Exo will degrade linear dsDNA starting from the 5’ end and generate 2 possible products: 1) a partially dsDNA duplex with single-stranded 3’ overhangs or 2) if the dsDNA was short enough, a ssDNA whose entire complementary strand was degraded.
Beta: Beta protects the ssDNA created by Exo and promotes its annealing to a complementary ssDNA target in the cell. Only Beta expression is required for recombineering with an ssDNA oligo substrate.
 |
|
Figure 1: Components of the Lambda Red Recombineering system. See text for details. |
Experimental outline
Below is a brief outline of a generic lambda red recombineering experiment (Figure 2). In the following sections, key steps that differ from traditional restriction enzyme cloning will be explained in greater detail.
- Substrate DNA design and generation
- Expression of lambda red recombination genes.
- Electroporation of substrate DNA and outgrowth of bacteria.
- Selection and confirmation of recombinant clones.
 |
|
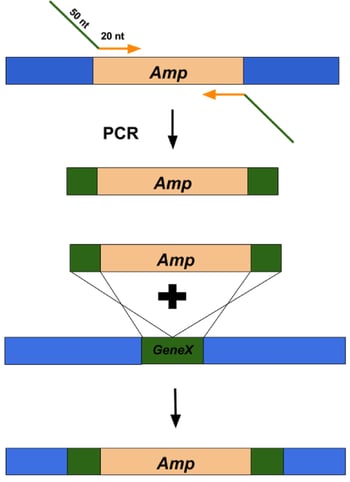
Figure 2: Overview of using Lambda Red recombineering system to replace a gene of interest with an antibiotic resistance cassette. |
Substrate DNA Design and Generation
Whether you use a linear dsDNA or ssDNA substrate depends on the goal of the experiment. dsDNA substrate is best for insertions or deletions greater than approximately 20 nucleotides, while ssDNA substrate is best for point mutations or changes of only a few base pairs.
dsDNA Substrate
dsDNA inserts can be made by PCR using primers that amplify the DNA sequence of interest and flank it with 50 base pairs of homology to the targeted insert site (Figure 2, top). These primers are typically ~70 nucleotides long (20 nucleotides that anneal to the DNA sequence of interest and 50 nucleotides of homology to regions flanking the target site). The best applications for dsDNA inserts include: large insertions or deletions, including selectable DNA fragments, such as antibiotic resistance genes, as well as non-selectable DNA fragments, such as gene replacements and tags. The typical frequency of recombinants is 1 positive clone out of 104 to 105 colonies.
 Pro Tip: Use a high-fidelity Taq polymerase with proof-reading ability for generating dsDNA recombineering substrates to avoid errors introduced during PCR.
Pro Tip: Use a high-fidelity Taq polymerase with proof-reading ability for generating dsDNA recombineering substrates to avoid errors introduced during PCR.
ssDNA Substrate
ssDNA substrates can be generated by ordering synthetic oligonucleotides or denaturing short PCR products. Either way, the substrate should be ~70-100 nucleotides long with the desired alteration(s) located in the center of the sequence. Since lambda red has a higher recombination frequency when the lagging strand of DNA is targeted, it’s best to determine the direction of replication through your region of interest and design an oligo that is complementary to the lagging strand. In practice, it’s easier to not determine the direction of replication and instead design oligos that target both strands. One of the two oligos will recombine with a higher efficiency than the other and you’ll find out empirically which is the lagging strand.
ssDNA substrate is more efficient than dsDNA with a recombination frequency between 0.1% to 1%, and can be increased to as high as 25-50% by designing oligos that avoid activating the methyl -directed mismatch repair (MMR) system. MMR’s job is to correct DNA mismatches that occur during DNA replication. There are two ways to avoid activating MMR: 1) use a strain of bacteria that has key MMR proteins knocked out or 2) specially design ssDNA oligos to avoid MMR:
1) E. coli with inactivated MMR: Using E. coli with inactive MMR is definitely the easier of the two options, but these cells are prone to mutations and will have more unintended changes to their genomes.
2) Designing ssDNA oligos that avoid MMR activation: There are two ways to design oligos that avoid MMR activation. The first way is to introduce a C/C mismatch at or within 6 base pairs of the edit site. This is an easy fix, but can only be used in limited circumstances. An alternative approach is to flank the desired change with 4-5 silent changes in the wobble codons, i.e. make changes to the third base pair of the adjacent 4-5 codons that alter the nucleotide sequence but not the amino acid sequence of the translated protein. These changes can be 5’ or 3’ of the desired change.
Table 1: Overview of ds- and ss-DNA Uses in Recombineering
| Substrate | Best Suited Application | Recombination Frequency | Special Considerations |
| dsDNA | Insertion or deletion of >20 nucleotides | 1 out of 104 to 105 | Use a high fidelity Taq polymerase to generate insert |
| ssDNA | Point mutations or changing a few nucleotides | 0.1-1% or 25-50% when activation of MMR is avoided | Introduce C/C mismatches or silent changes in wobble codons to avoid MMR activation |
Expression of Lambda Red recombination genes
There are four ways to express the lambda red recombineering system: 1) From a bacteria with integrated defective prophage 2) from a plasmid, 3) from mini-λ or 4) from the lambda red phage itself (each of these will be discussed in more detail below). Controlling expression of Red proteins is critical for minimizing the toxic effects of Gam expression and to limit spontaneous mutations that occur when Red is constitutively expressed. Which recombination system you use depends on what type of DNA you want to edit; however, BAC DNA can be modified with any of the below described approaches.
Bacterial strain with integrated defective prophage
A number of E. coli strains exist that stably express lambda red recombineering genes due to integration of a defective lambda red phage. One such strain is DY380, which is derived from the DH10B E. coli strain. Several other bacterial strains commonly used for recombineering can be found in Thomason et al and Sharan et al (a few strains can also be found at Addgene, but be sure to read the associated papers for their particular uses - *note* not all linked strains contain the Lambda Red system).
In some of these strains, expression of exo, beta and gam is tightly regulated by the endogenous phage promoter pL and repressor CI. For recombineering purposes, a temperature sensitive version of the repressor gene, CI857, is used. This mutant repressor prevents expression of the recombination genes at low temperatures (30-34˚C). Shifting bacteria to 42˚C for 15 minutes quickly inactivates the repressor and allows for expression of the recombination genes. After this, lowering the temperature allows the repressor to re-nature and again repress expression of exo, beta and gam. A major advantage of using this method for lambda red expression is that it doesn’t require antibiotic selection to maintain expression of the recombineering system. This setup can be also be used to modify chromosomal genes. After the initial editing event, the defective prophage can be removed from the chromosome of the host E. coli by a second lambda red recombination event. Alternatively, if the modified allele is selectable, it can be transferred to a different genetic background via P1 transduction.
Plasmid
Expressing lambda red genes from a plasmid allows for a mobile recombineering system, but tight regulation of expression is required for a successful experiment. Promoters commonly used to control expression of Red include the IPTG-inducible lac promoter, the arabinose-inducible pBAD promoter and the endogenous phage pL promoter. It is a good idea to choose plasmids that also express the repressors associated with these promoters (lacI, araC, cI857) to limit leaky expression of the Red system. Using a plasmid to express the lambda recombineering system is best suited for editing bacterial chromosomal DNA because it is easy to remove the recombineering system once recombinant clones are generated. A simple way to do this is to express lambda red genes from a plasmid with a heat sensitive origin of replication. Once the recombineering system is no longer needed, bacteria can be “cured” by growing them at 42℃.
Mini-λ
A hybrid between using a plasmid and stable integration of a defective prophage is to use mini-λ, a defective non-replicating, circular piece of phage DNA, that when introduced into bacteria, integrates into the genome. Mini-λ uses the endogenous red promoter pL and the cI857 repressor to regulate expression. An antibiotic can be used to select for positive clones but, because mini-λ stably integrates, drug selection is not required for maintenance. A temperature shift to 42˚C not only allows for activation of the red genes needed for recombineering, but also leads to expression of the int and xis genes which are responsible for excision of mini-λ from the host’s chromosome. After this, mini-λ can readily be purified from bacteria just like a plasmid.
Phage
A final option for expressing the Red system is to use a lambda red phage, λTetR, that carries the tetracycline resistance gene and the lambda red repressor cI857. Once introduced, the prophage is stable and no longer requires drug selection. One drawback to this approach is that it requires the generation of bacteriophage, which is not a common molecular biology technique. However, an advantage of this method is that you can stably integrate the Red system into your own strain of interest and P1 transduction could be used to move the modification into a different background, if needed. This approach is best suited for modifying plasmids or BACs because it results in stable integration of the phage into the E. coli genome.
Selection and confirmation of recombinant clones
If a antibiotic resistance gene has been inserted, recombinants can first be selected via antibiotic resistance, but all clones should be further tested to confirm the presence of the desired modification. Colony PCR can be used to screen for positive clones in most cases, and restriction enzyme digest can be used to screen plasmids for the appropriate mutations. Point mutations and other subtle changes should be confirmed by sequencing, which is also the best final confirmation for all clones, regardless of what type of DNA is being targeted for modification: the E. coli chromosome, a plasmid, or a BAC.
Pro Tip: ssDNA substrates have a much higher rate of recombination than dsDNA substrates, so you should screen more clones when using dsDNA.
Have you used the lambda red recombineering system? Share your tips below!

Many thanks to our guest blogger, Beth Kenkel
 Beth Kenkel is currently a research scientist in the Department of Laboratory Medicine at the University of Washington. She is particularly interested in science communication and in vitro diagnostics.
Beth Kenkel is currently a research scientist in the Department of Laboratory Medicine at the University of Washington. She is particularly interested in science communication and in vitro diagnostics.
References
1. Costantino N, Court DL. Enhanced levels of λ Red-mediated recombinants in mismatch repair mutants. Proceedings of the National Academy of Sciences of the United States of America. 2003. doi:10.1073/pnas.2434959100. PubMed PMID: 14673109. PubMed Central PMCID: PMC307639.
2. Daiguan Yu, Hilary M. Ellis, E-Chiang Lee, Nancy A. Jenkins, Neal G. Copeland, and Donald L. Court. An efficient recombination system for chromosome engineering in Escherichia coli. PNAS 2000 97 (11) 5978-5983; May 16, 2000, doi:10.1073/pnas.100127597. PubMed PMID: 10811905.PubMed Central PMCID: PMC18544.
3. Murphy, K. 2016. λ Recombination and Recombineering, EcoSal Plus 2016. doi:10.1128/ecosalplus. PubMed PMID: 27223821.
4. Sharan SK, Thomason LC, Kuznetsov SG, Court DL. Recombineering: A Homologous Recombination-Based Method of Genetic Engineering. Nature protocols. 2009;4(2):206-223. doi:10.1038/nprot.2008.227. PubMed PMID: 19180090. PubMed Central PMCID: PMC2790811.
5. Thomason, L. C., Sawitzke, J. A., Li, X., Costantino, N. and Court, D. L. 2014. Recombineering: Genetic Engineering in Bacteria Using Homologous Recombination. Current Protocols in Molecular Biology. 106:V:1.16:1.16.1–1.16.39. PubMed PMID: 24733238.
6. National Cancer Institute Recombineering Pages.
Additional Resources on the Addgene Blog
- Read about Homology Directed Repair
- Use Knockout/Knockin Plasmids to Disrupt Genes in Mammalian Cells
- CRISPR Methods for Bacterial Genome Engineering
Resources on Addgene.org
- Browse our Microbiology Pages
- Find Lambda Red Plasmids and Strains
- Check Out Other Molecular Biology Protocols
Topics: Plasmid Cloning, Plasmids





Leave a Comment