One of the most powerful strategies to investigate a gene's function is to inactivate, or "knockout", the gene by replacing it or disrupting it with an piece of DNA designed in the lab. Specially constructed plasmids can be used to replace genes in yeast, mice, or Drosophila through homologous recombination. The concept is simple: deliver a template with a modified version of the targeted sequence to the cell which will recombine the template with the endogenous gene. Here, we'll describe the techniques and the plasmids used to inactivate specific genes in mammalian cells. Despite the popularity of CRISPR-based knockout/knock-in systems, these systems remain valuable, especially in cases where CRISPR cannot be used (e.g. there are no suitable PAM sequences nearby or your gene of interest is difficult to target specifically with a gRNA). Be sure to keep these techniques in mind when choosing a knockout strategy!
Knockout plasmids
Homologous recombination is a mechanism to accurately repair harmful double stranded breaks, in which nucleotide sequences are exchanged between two similar or identical molecules of DNA. Gene targeting takes advantage of this natural process to replace a targeted genetic locus with homologous sequence using a specially designed vector that contains sequence homology to the locus of interest. To give you an idea of the process, we’ll walk through an experiment designed to knock out exon 2 of a given gene.
 |
|
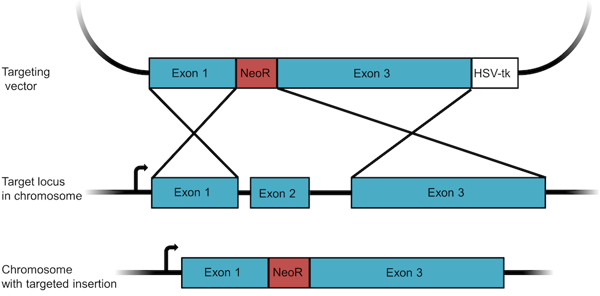
Figure 1: A knockout targeting vector designed to insert a resistance gene. The vector contains a neomycin resistance gene (NeoR) flanked by homology arms. The negative selection marker HSV-tk is used to select against random recombinants. |
- Design your targeting construct. For recombination to occur in a cell, a minimum of 2 kb of sequence homology is required, but 6 to 14 kb of homology is typical for targeting constructs. In the example shown in Figure 1, large sequences corresponding to exons 1 and 3 of the targeted gene have been cloned into the vector on either side of an antibiotic resistance gene. To avoid selecting for cells in which the construct has randomly integrated into the genome, a negative selection marker like the HSV thymidine kinase (HSV-tk) is included just outside one of the homology arms. When we select cells later, we will first perform a positive selection for the antibiotic resistance gene and then a counter selection for the negative selection marker - this latter step will kill off many of the cells that have randomly integrated all or large portions of the plasmid.
- Deliver your construct to your target cells. After recombination, exon 2 of the targeted gene will be removed from the chromosome and and replaced by the resistance gene.The gene is thus disrupted, or knocked out.
- Use positive and negative selection to find correctly recombined cells. Recombination is a rare event, so you must select as opposed to screen for cells where recombination has occurred. Neomycin, puromycin, and hygromycin resistance genes are commonly used for positive selection. While the positive selection marker selects for recombination, the negative selection marker selects against improper, random recombination into a different locus. Correctly recombined cells will not contain the negative selection marker, but cells with random recombination may incorporate the negative selection marker into the genome. This final product of non-homologous recombination can survive positive selection, but it will be sensitive to negative selection.
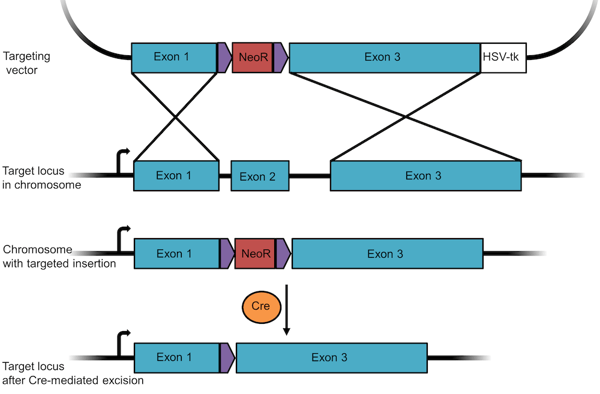
- Remove the positive selection marker. Since an antibiotic resistance gene can affect a cell's phenotype, researchers usually remove it after selection using the Cre/Lox recombination system. After “floxing” the resistance gene through the insertion of flanking LoxP sequences, you can remove the gene through the addition of Cre recombinase (Figure 2).
 |
|
Figure 2: A knockout targeting vector designed to insert and later remove a resistance gene. In this construct, the neomycin resistance gene is floxed so that it can be removed post-selection through Cre-mediated recombination. |
Knockout/Knock-in plasmids
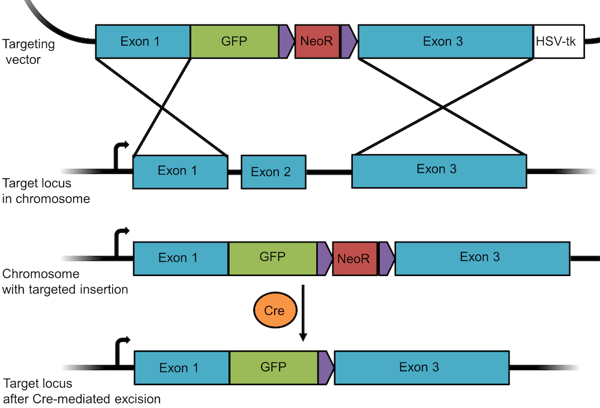
Gene targeting methods also make it possible to insert, or knock-in, any gene, tag, or mutated exon into the genome. For this purpose, the sequence to be inserted is cloned into the vector between the homologous sequences together with the positive selection marker. In order to both knockout a given gene and insert GFP into the genome, we’d create a plasmid similar to the one shown below, where the sequence of GFP is cloned together with the Neomycin resistance (NeoR) gene between exon 1 and 3 of the targeted gene. Upon recombination, the GFP/NeoR cassette is inserted in place of exon 2. Thus, the targeted gene is disrupted (knocked out) but the inserted GFP is expressed (knocked in). As seen in the above example, you can remove a floxed resistance gene using Cre recombinase. If GFP is under control of an endogenous promoter, you can use expression GFP to track cells participating in development or other physiopathological events to which the chosen promoter responds. You can also use this method to tag an endogenous protein with GFP, as seen in blue flame plasmid OCT4-eGFP-PGK-Puro from the Jaenisch lab.
 |
|
Figure 3: A knockout/knock-in targeting vector designed to insert GFP into a given locus. |
Conditional knockouts
The methods and plasmids described in the preceding sections are simple ways of knocking out a non-essential gene of interest. However, if your gene of interest is essential, a true knockout can be lethal, and you'd instead want to create a conditional knockout.
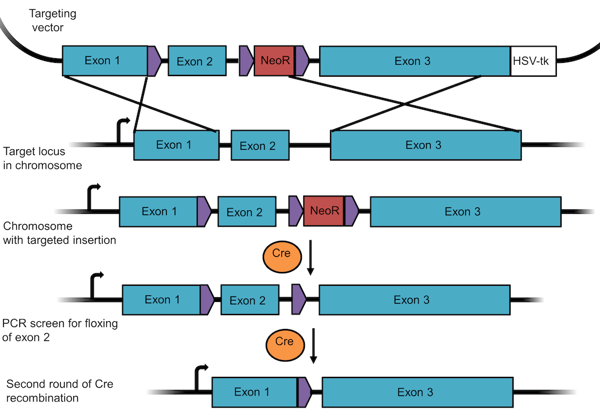
To make a conditional knockout, researchers often use the Cre/Lox system described earlier. In this case, you design your targeting vector such that a set of three LoxP sites flank the resistance gene and the targeted exon in the gene of interest (Figure 4). When recombination occurs, the gene still functions normally because one of its exons has simply been replaced with the same sequence flanked by LoxP sites while the resistance cassette has been placed into an intron.
 |
|
Figure 4: A targeting vector to create a floxed allele for a conditional knockout. This experimental design requires two rounds of recombination – the first removes the selection marker, the second the floxed exon. |
After recombination has occurred, you'll first remove the resistance marker using Cre recombinase. Since there are 3 loxP sites, recombination can occur in multiple ways. The desired recombination event will remove NeoR only and leave exon 2 floxed, as seen in line 4 of Figure 4. Since the loxP sites are located in intronic regions, this gene will still be expressed. You'd first screen for this specific recombination outcome using PCR and then generate a monoclonal cell line with the floxed exon. You can then conditionally remove this exon (and hence knock out the gene) through a second round of Cre recombination.
The future of knockout plasmids
Although these methods have been used to create many knockout cell lines and animal models, their efficiency is very low, ranging from undetectable to 0.1%. In contrast, new genome editing techniques such as CRISPR are easier to use and are more efficient at inactivating genes. CRISPR can target a genomic sequence and create a break that can be repaired by homologous recombination using a repair template. These templates can include loxP sites to create conditional floxed alleles. Although CRISPR is very good at making knockouts, knocking in large sections of DNA can be more difficult. Addgene depositors have developed new CRISPR-based methods for various knock-ins, discussed in our CRISPR 101 eBook.
It’s hard to believe that the first knockout mouse was created in 1989, less than thirty years ago. As traditional knockout and new CRISPR tools continue to be perfected, the production of cells and mouse knockout lines should increase. New genome engineering tools also offer hope for the creation of new knockout animal models in species that were previously difficult to engineer, like rats. If you have tools for creating knockout animals or cell lines, please consider sharing them by depositing with Addgene!
Figures were adapted from: Hall B, A Limaye, & AB Kulkarni. (2009) "Overview: Generation of Gene Knockout Mice". Curr Protoc Cell Biol. 19: 1217. PubMed PMID: 19731224 PubMed Central PMCID: PMC2782548
Additional Resources on the Addgene Blog
- Get the Basics of Homology Directed Repair
- Read about Generating Knockouts with CRISPR through Non-Homologous End Joining
- Learn How the FLEx Switch Can Be Used to Turn Genes On and Off
Resources on Addgene.org
- Find Mammalian Expression Vectors
- Find Cre-Lox Plasmids
- Learn More about CRISPR

Topics: Plasmids 101, Cre-lox, Plasmids





Leave a Comment