This post was contributed by Scott Findlay, a Postdoctoral Fellow at the University of Alberta.
If you’re like many researchers these days, you are ready to take (if you haven’t already) the plunge into the world of precision genome editing. When it comes time to (hopefully) validate successful mutation of your favourite gene, there are several different methods available. Thankfully, there are many great resources available to help guide you through the rough waters of mutation validation, such as this "CRISPR 101" post. Next-generation sequencing technologies are the gold standard but they remain cost-prohibitive for many labs, and are often impractical for small projects. Most researchers instead turn to so-called “mismatch nuclease” assays (e.g. Surveyor® or T7E1) for mutation detection. While these methods paved the way for mutation validation, we found these assays frustrating to work with, time consuming, and minimally informative. In this blog post, we’ll introduce digital PCR as an emerging validation technology. Digital PCR has several advantages over mismatch nuclease assays that will be elaborated below.
Digital PCR
Digital PCR (dPCR) involves splitting up a sample into thousands of physically isolated partitions (such as droplets or wells on a chip), such that most partitions will contain either one or no copies of the target DNA. Conventional thermal cycling is then performed to amplify the target using PCR. Instead of tracking the reaction progress as in real time PCR, each reaction partition is individually assessed for a positive or negative (digital) signal at the end of the reaction (Figure 1). Partitions that did not contain any target DNA at the onset of PCR are negative, whereas those that did are positive. Since there are many thousands of partitions, statistical modelling can be used to determine the actual number of target DNA molecules present in the original sample with a great deal of precision. This means digital PCR provides an absolute measure of target abundance, does not require standard curves, and can theoretically detect a single target molecule in a sample.
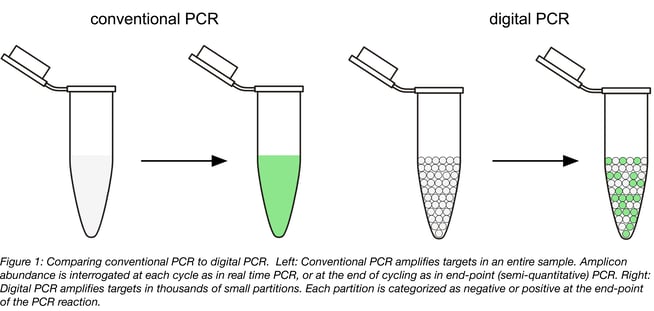
Digital PCR assays for mutation screening
Now that we have introduced how digital PCR works, let’s explain what kind of assays can be used to sniff out your desired mutations. The assays you will use aren’t that much different from conventional duplexed primer probe assays often used in real time PCR. These assays can be designed to detect either a donor sequence integrated through homology-directed repair (HDR) (1), or indel mutations resulting from non-homologous end joining (NHEJ) (2-5), depending on your desired genome edit.
Since exploitation of NHEJ to generate translational reading frame-altering indels for “functional knockout” of a gene of interest is one of the most common applications of precision genome editing, the remainder of this post will focus on assays to detect NHEJ. These assays include a forward and reverse primer to amplify the target locus, as well as a reference probe designed to bind at an unedited site distal tothe predicted double strand break, and a NHEJ/ “drop-off” probe designed to bind directly at the predicted double strand break site (Figure 2A).
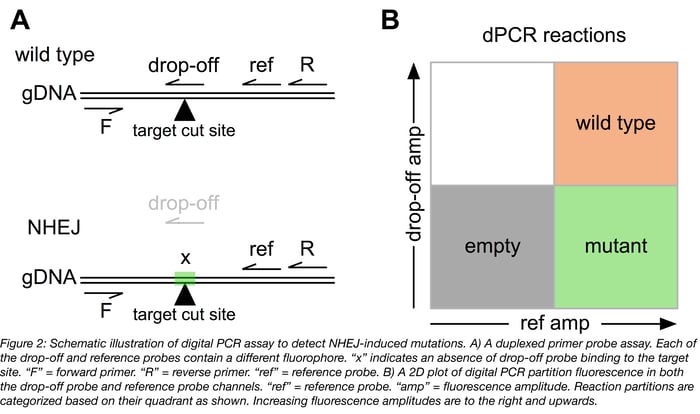
The single molecule resolution of digital PCR allows classification of individual target molecules based on their mutation status. The drop-off probe is designed to perfectly complement the wild type sequence and is not capable of binding a mutated target. The amount of both mutated and wild type target in each sample can be quickly calculated (details in [2]) and can be used to quantify rates of mutation in a bulk or single cell-derived genomic DNA sample as part of a precision genome editing workflow.
Digital PCR comparison to other validation methods
As mentioned, these assays have several major advantages over the commonly employed mismatch nuclease assays.
- Sensitivity: Less than 20 ng of total gDNA is sufficient for analysis. We have routinely successfully analyzed gDNA from a few thousand cells from single wells of a 96-well plate without prior quantification. Comparatively, mismatch assays require between 200 and 500 ng of purified PCR product.
- Low detection limits: Digital PCR assays can accurately quantify low levels of mutant target (as low as 20 pg or 0.02%) in a high background of wild type targets. Comparatively, detection limits of about 5% mutant targets have been reported for mismatch nuclease assays (2, 6, 7).
- Ability to distinguish between mono-allelic and bi-allelic mutations: Perhaps the most noteworthy advantage of digital PCR assays is that they are easily able to distinguish single-cell derived samples with mono-allelic mutations versus those with the bi-allelic mutations typically desired when screening clones for complete functional knockout of your target gene. Mismatch nuclease assays are completely “blind” to this very important difference (Figure 3).

If you are interested in harnessing the power of digital PCR screening to validate your genome edits, this paper provides detailed instructions on how to design such assays, in addition to testing their performance (2). Remember that although digital PCR is a great screening tool, it still requires target DNA sequencing to confirm the exact nature of your mutations. A great description of the plethora of available sequencing options can be found here. Good luck with your genome editing, and may all your NHEJ mutations be out of frame!
 Scott Findlay is currently a Postdoctoral Fellow at the University of Alberta. He is interested in everything molecular biology. Follow him on Twitter @AnotherLabRat.
Scott Findlay is currently a Postdoctoral Fellow at the University of Alberta. He is interested in everything molecular biology. Follow him on Twitter @AnotherLabRat.
References
1. Miyaoka, Y. et al. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat Meth 11, 291–293 (2014). PubMed PMID: 24509632. PubMed Central PMCID: PMC4063274.
2. Findlay, S. D., Vincent, K. M., Berman, J. R. & Postovit, L.-M. A Digital PCR-Based Method for Efficient and Highly Specific Screening of Genome Edited Cells. PLoS ONE 11, e0153901–17 (2016). PubMed PMID: 27089539. PubMed PMCID: PMC4835065.
3. Mock, U., Hauber, I. & Fehse, B. Digital PCR to assess gene-editing frequencies (GEF-dPCR) mediated by designer nucleases. Nat Protoc 11, 598–615 (2016). PubMed PMID: 26914317.
4. Mock, U. et al. mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Research 43, 5560–5571 (2015). PubMed PMID: 25964300. PubMed Central PMCID: PMC4477672.
5. Sedlak, R. H. et al. Digital detection of endonuclease mediated gene disruption in the HIV provirus. Sci. Rep. 6, 20064 (2016). PubMed PMID: 26829887. PubMed Central PMCID: PMC4735761.
6. Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology 31, 822–826 (2013). PubMed PMID: 23792628. PubMed Central PMCID: PMC3773023.
7. Vouillot, L., Thélie, A. & Pollet, N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5, 407–415 (2015). PubMed PMID: 25566793. PubMed Central PMCID: PMC4349094.






Leave a Comment