Since the discovery of GFP over 50 years ago, the growing spectrum of fluorescent proteins (FPs) has been an invaluable resource for studying the organization and function of cellular systems. FPs have been used to track protein localization, cell structure, intracellular trafficking, and protein turnover rates. Additionally, by engineering FP fusions associated with cellular organelles, scientists have been able to study many cellular processes, including mitosis, mitochondrial fission/fusion, nuclear import, and neuronal trafficking. Although FPs have enabled discovery of many cellular mechanisms, there are some limitations to working with FPs. Overexpression of fluorescently tagged proteins can lead to improper protein localization, protein aggregation, or disruption of normal protein function, and ultimately misinterpretation of the protein’s cellular role.
One way to avoid the pitfalls of overexpressed fluorescent protein fusions is to replace the genomic copy of your gene of interest with a fluorescent protein fusion. CRISPR genome editing can help accomplish this goal. The power of this system lies in the ability of the endonuclease Cas9 to create a DNA double stranded break (DSB) at a genomic site specified by a guide RNA (gRNA) sequence. Users can design gRNAs to induce the break at a specific genomic site and, using the endogenous homology directed repair pathway, a new user-defined DNA sequence (like GFP) can be inserted at the DSB. CRISPR has allowed scientists to tag and light up endogenous genes of interest to better understand normal protein function. However, CRISPR protein tagging does have its limitations, especially when trying to perform high-throughput experiments. If a user wants to carry out a high-throughput genetic screen, it is expensive and time consuming to individually generate gRNAs and homology arm repair templates containing the tag for each insertion. Also, it remains challenging to screen for the genetically modified strains that contain your newly tagged gene. Most insertions can only be detected by labor-intensive processes like PCR or by evaluating visual phenotypes.
Improving C. elegans fluorescent protein tagging with SapTrap
The Jorgensen Lab recently developed a modular plasmid assembly toolkit called SapTrap to improve CRISPR genomic tagging in C. elegans. SapTrap offers scientists a convenient one tube assembly reaction to generate high-throughput targeting vectors. These vectors can be used to tag endogenous genes and simultaneously introduce a selection marker for screening modified strains. With SapTrap, the user first designs either oligos or synthetic DNA for the desired gRNA target sequence, as well as the 5’ and 3’ homology arm repair template (Fig. 1, Step 1). There is no need for PCR or cloning, as digestion of the destination vector with SapI yields 2 sites- the first site accepts the sgRNA target sequence for U6 promoter expression and the second site accepts the homology arm repair template.
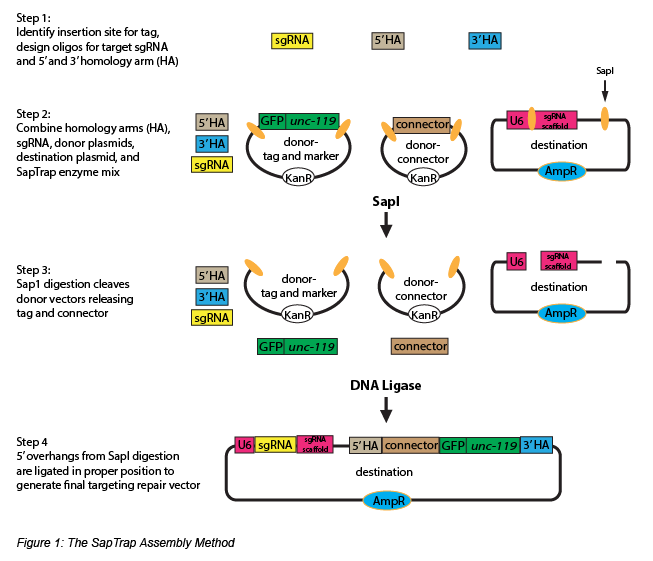
SapTrap includes a prebuilt donor plasmid library containing several types of fluorescent (EGFP, tagRFP, mCherry) and nonfluorescent (Halo, SNAP) tags, a selectable marker (floxed Cbr-unc-119) for easy screening of the insertion event, and a variety of connector modules (linker sequences between the tag and homology arms). Digestion of the donor plasmids with SapI allows the tag, selectable marker, and connector to be released (Fig. 1, Step 2-3). Since donor plasmids of the same type will produce the same unique SapI 5’ overhang, different combinations of connector and tag donor plasmids from the library can be used to generate functionally unique repair templates. A final ligation reaction correctly assembles the final targeting vector (Fig. 1, Step 4), and co-injection of the targeting vector and a Cas9 expression plasmid will insert the desired genetic tag and marker sequence into the targeted loci. The selectable marker can be removed by Cre-mediated excision for scarless tag insertion.
In addition to the obvious advantages SapTrap offers for tagging a single genetic locus, the toolkit also offers repair templates for tagging proteins in a tissue specific manner, as well as 3-site destination vectors for inserting a tag at multiple target sites. SapTrap vectors can be easily modified to tag hundreds of genes, generating powerful genetic screening libraries.
Additional protein tagging systems
For those who do not work with C. elegans, several groups have designed modular toolkits to assist with tagging genomic loci in other model systems, including mammalian cells.
- CRISPaint (CRISPR-assisted insertion tagging), developed by the Hornung Lab, allows users to easily create C-terminal tag fusions of endogenous genes in human cells. To use CRISPaint, users need 3 vectors: 1) a gRNA vector that targets the gene of interest, 2) a plasmid to specify the reading frame of the insertion, and 3) a vector containing the desired tag, which can be obtained as a universal donor plasmid. The CRISPaint toolkit includes several donor plasmids for protein tagging, including luciferase (NanoLuc), fluorescent proteins (TagGFP2, TagBFP, TagRFP, and T2A-TurboGFP-PEST), and small epitope tags (HA, Myc, Strep tag II, AviTag, HaloTag, SpyTag).
- The Foerstemann lab developed a polymerase chain reaction (PCR)-based system that allows users to generate a plasmid containing a direct fusion of the desired gRNA to the U6 promoter, and a second plasmid containing the homology arms and an N- or C-terminal tag. The two PCR reactions are then mixed and transfected along with a Cas9 expression vector. They can also be introduced directly into a Drosophila S2 cell line stably expressing Cas9. This PCR toolkit offers C- and N-terminal tagging vectors (eg GFP, Flag, YFP, Strep, TEV-V5) with either blasticidin or puromycin selection.
- Researchers at the Allen Institute for Cell Science recently developed a collection of plasmids to fluorescently tag markers of cellular structures in mammalian cells. These plasmids use fluorescent proteins flanked by long regions of homology to the gene of interest to promote homology directed repair after a CRISPR/Cas9 induced break. Learn more about these constructs and the cell lines they’ve been used to create in the Allen Institute’s recent blog post.
- For more information on other CRISPR/Cas systems used for endogenous protein tagging, please visit our website CRISPR/Cas Plasmids - Protein Tagging.
Beyond simply monitoring protein localization and activity, CRISPR tagging systems can be modified to assay additional protein characteristics. For example, Masato Kanemaki's lab has developed a CRISPR/Cas-based system for tagging endogenous proteins with an auxin-inducible degron (AID) tag to generate proteins which can be rapidly and reversibly degraded after the addition of auxin to the culture medium. Any number of regulatory sequences, including protein enhancers and repressors, can be included in the repair template for inducible alteration of protein expression levels.
CRISPR tagging systems offer users an easy and efficient way to tag endogenous proteins for studying protein expression, localization, and protein-protein interactions. Additionally, thanks to the modular arrangement of vectors included in tagging systems like SapTrap, scientists will be able to generate genome-wide libraries containing fluorescently-tagged proteins at endogenous loci, shedding light on previously unknown protein functions.
References
Schwartz, Matthew L. and Eric M. Jorgensen. “SapTrap, a Toolkit for High-Throughput CRISPR/Cas9 Gene Modification in Caenorhabditis elegans.” Genetics. 202(4) (2016):1277-88. PubMed PMID: 26837755. PubMed Central PMCID: PMC4905529.
Schmid-Burgk, Jonathan L., et al. “CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism.” Nat Commun. 7 (2016):12338. PubMed PMID: 27465542. PubMed Central PMCID: PMC4974478.
Natsume, Toyoaki, et al. “Rapid Protein Depletion in Human Cells by Auxin-Inducible Degron Tagging with Short Homology Donors.” Cell Rep.15(1) (2016):210-8. PubMed PMID: 27052166.
Kunzelmann, Stefan, et al. “A Comprehensive Toolbox for Genome Editing in Cultured Drosophila melanogaster Cells.” G3 (Bethesda) 6(6) (2016):1777-85. PubMed PMID: 27172193. PubMed Central PMCID: PMC4889673.
Additional Resources on the Addgene Blog
- Learn about Fluorescent Protein Timers
- Check out Photoactivatable Fluorescent Proteins
- Use Rosella to Monitor Autophagy
Resources on Addgene.org
- Check out the SapTrap Kit Page
- Find Other Fluorescent Protein Kits
- Browse the Fluorescent Protein Collection
Topics: Fluorescent Proteins, Generating Fusions





Leave a Comment