This post was contributed by guest blogger Pooran Dewari, a postdoc in Steve Pollard’s lab at the MRC Centre for Regenerative Medicine (CRM), Edinburgh.
Most commercial antibodies do not work in pull-down assays: Epitope tagging provides a solution
Proteins - the workhorses of the cell – never work alone in the cellular milieu. It is, therefore, critical to understand how proteins interact with one another (or with DNA) to perform diverse biochemical tasks in the cell. One of the most popular approaches to study protein interactions is the pull-down assay, wherein a protein of interest can be captured along with its associated partners. Common pull-down assays include immunoprecipitation mass spectrometry (IP/MS) and chromatin immunoprecipitation (ChIP). In IP/MS, a target protein is first immunoprecipitated - along with its associated protein complexes - from the cell-lysate using antibodies against the target protein. The captured protein complexes are then analysed by mass spectrometry to identify the interacting proteins. Similarly, in ChIP-seq assays, chromatin fragments that are bound by a protein of interest are pulled-down and later coupled to high-throughput sequencing to identify genome-wide binding patterns of the target protein.
Both IP/MS and ChIP-seq methods provide a wealth of information about how protein networks work in cells. A major bottleneck, however, to these IP-based assays is to find (or generate in-house) a pre-validated antibody for each target protein. To study 20,000 human proteins, this would be time-consuming, expensive, and not to mention frustrating. One elegant solution to sidestep the antibody problem is to tag the protein of interest with a small biochemical “epitope tag” such that these tagged proteins can be immunoprecipitated using a standard tag-specific antibody (Jarvik and Telmer 1998). Indeed, this method can be used at high throughput because numerous cell lines can be generated, each with a different tagged protein of interest, and a universal all-purpose “tag antibody” can be used for downstream IP/ChIP assays.
Creating epitope-tagged clonal cell lines remains a challenge as it relies on inefficient homology-directed repair (HDR) to correct double-strand breaks (DSBs). DSB repair by HDR facilitates in-frame insertion of tag-encoding sequences at the intended target site. With the widespread adoption of CRISPR/Cas9 technology and rapid improvements in the field, it has recently become possible to introduce DSBs and concomitant insertion of epitope tags at target genes in mammalian cells (Bressan et al. 2017; Liang et al. 2017).
Current methods of epitope tagging, however, suffer from many pitfalls: they require construction of plasmid vectors and/or rely on cumbersome in vitro synthesis of single guide RNAs (sgRNAs), are time-consuming, have been typically optimised for immortal/genetically-corrupt cell lines, lack scalability, and have low knock-in efficiency. Together, these factors prevent stem cell researchers from generating clonal knock-in lines, especially for ambitious projects that require epitope tagging of a large cohort of genes (e.g. tagging genes representing a protein super family).
To overcome these challenges, we set out to develop a hassle-free, highly efficient Cas9 protein-based method for epitope tagging in mammalian stem cells. For easy scalability to the 96-well format, we developed a “TAG-IN” bioinformatics tool for guide RNA and donor DNA design, and implemented our optimised protocols into a medium-throughput pipeline for epitope tagging. Using our 96-format pipeline, we can routinely achieve 5-30% tag knock-in efficiency across different stem cell types. This has facilitated easy generation of clonal knock-in lines that we now use for IP and ChIP assays.
Tag 96 genes at a time using Custom Synthetic cr:tracrRNA/Cas9 Ribonucleoprotein (csRNP) delivery
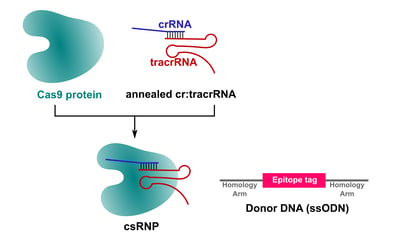
Epitope tagging of mammalian genes using CRISPR depends on successful delivery of three reagents to cells: a guide RNA, Cas9 protein, and a donor DNA that contains the tag sequence flanked by homology arms (Figure 1). In the first step of optimization, we started off by using custom synthetic two-part cr/tracrRNAs for CRISPR experiments (in collaboration with Integrated DNA Technologies, USA). These synthetic RNAs can also be purchased from various other suppliers with a typical turnaround time of 5-7 days. The crRNA and tracrRNA are annealed on a PCR block and the annealed cr:tracrRNA is then mixed with recombinant Cas9 protein to form the ribonucleoprotein complex (csRNP, Figure 1). We use in-house purified Cas9 protein, which, in our hands, is both cost-effective and highly active. After csRNP complex formation, a single-stranded donor DNA (ssODN) that contains the tag-encoding sequence and short homology arms (60-70 bp) is added to the complex. This complete transfection mix, i.e. csRNP plus ssODN, is then transfected into stem cells. Cells are allowed to recover for 2-5 days and then analyzed for tagging efficiency using standard immunocytochemistry (ICC) assays.
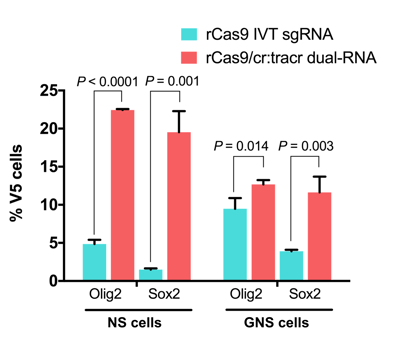
To calculate the tagging efficiency in the transfected cells, we count the number of tag-positive cells (tag ICC) as a percentage of total cells in the field (DAPI). Using the ICC assay as a direct readout of tag insertion efficiency, we have recently demonstrated that the two-part cr:tracrRNA system is much more potent than the in vitro-transcribed single guide RNA (IVT sgRNA) approach (Dewari et al. 2018 and Figure 2). Not only is the cr:tracrRNA approach highly efficient, it is also straightforward as the RNA molecules can be directly purchased and there is no need to construct plasmid vectors or synthesize/purify IVT sgRNA in the lab.
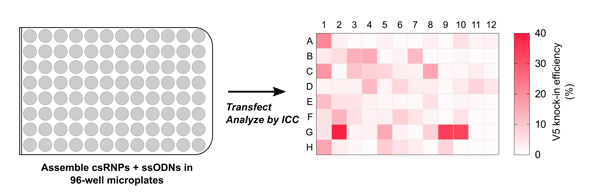
Encouraged by the high efficiency and easy procurement of cr:tracrRNA reagents, we scaled up these tagging experiments by assembling csRNPs in a 96-well plate format and tested parallel tag knock-in for 96 different genes in a single go (Figure 3). We were able to tag >30 genes/plate in the first shot with typical knock-in efficiencies of 10-20%. Together, our results show that genes can be tagged with high knock-in efficiency in the 96-well format and our csRNP method suits both small and medium-throughput epitope-tagging experiments. Notably, we could tag >60 transcription factors in a short (3-week) time window; this was possible because reagents could be purchased directly from commercial suppliers without investing time and labour on reagent preparation in the lab.
How to set up csRNP knock-in experiments
If your lab is new to CRISPR/Cas9 experiments, there is no need to worry at all. Setting up tag knock-in experiments using the csRNP method is as simple as doing routine PCR assays. Here is a simple to-do-list to follow when planning epitope tagging experiments:
- Retrieve Ensembl Transcript ID(s) of your favourite gene(s) from Ensembl genome browser: https://www.ensembl.org/index.html
- Design crRNA/ssODN: Using Ensembl Transcript ID(s) as an input query, pick crRNA (guide RNA sequence targeting the gene of interest) and ssODN (tag-encoding donor DNA) using our TAG-IN bioinformatics tool.
A recent article by Clarke et al shows that crRNAs binding to the template strand (strand used by RNA polymerase II for transcription) are more effective for genome-editing events. In the light of these results, we re-analyzed our data and found that 75% of our highest knock-in efficiency CRISPR reagents had crRNAs that bind to the template strand. Therefore, I suggest that picking a crRNA complementary to the template strand could increase knock-in efficiency. Note: Our TAG-IN tool can compute crRNA/ssODN for up to 95 genes in less than a minute. As of now, the tool is available for human and mouse genomes only and deals with C-terminus epitope tagging. If you work with other model systems, you will have to design your crRNAs using other web-based design tools (for C-terminus tagging, pick a crRNA that cuts 8-15 bp downstream of Stop codon; for N-terminus tagging, a crRNA cutting 8-15 bp upstream of your gene’s Start codon will be suitable). For N-terminus tagging, the promoter region of both the alleles should be sequence-confirmed for "indel-free" knock-in. Another good design tool can be found at Desktop Genetics .
Note: Our TAG-IN tool can compute crRNA/ssODN for up to 95 genes in less than a minute. As of now, the tool is available for human and mouse genomes only and deals with C-terminus epitope tagging. If you work with other model systems, you will have to design your crRNAs using other web-based design tools (for C-terminus tagging, pick a crRNA that cuts 8-15 bp downstream of Stop codon; for N-terminus tagging, a crRNA cutting 8-15 bp upstream of your gene’s Start codon will be suitable). For N-terminus tagging, the promoter region of both the alleles should be sequence-confirmed for "indel-free" knock-in. Another good design tool can be found at Desktop Genetics .
For ssODN design, remember to incorporate PAM-blocking mutations (e.g. convert the NGG PAM to NGC in your repair template) and use homology arms of 60 -70 bp . - Purchase gene-specific crRNAs and matched ssODNs from commercial suppliers of CRISPR reagents (turnaround time 5-7 days); because the tracrRNA is the same in all csRNP experiments, it can be ordered in bulk and stored in small aliquots at -80 oC.
- Assemble csRNP: Anneal crRNA and tracrRNA using a PCR block. Add Cas9 protein to the annealed cr:tracrRNA and incubate at room temperature for 10 min.
- Deliver csRNP into cells: Prepare cells for transfection, add csRNP and ssODN to the cell suspension, transfect and let cells recover for 2-5 days.Note: We use the Nucleofection method for delivery of csRNP reagents into cells. Optimization of delivery methods (electroporation, lipofection etc) is the most crucial aspect of knock-in experiments. You’ll need to optimize csRNP delivery for each cell type you work with (optimization includes selection of transfection buffer and electroporation pulse program to achieve high transfection efficiency and good cell-viability).
- Analyse tag knock-in success by ICC assays using a tag-specific antibody.Note: Always include a positive and a negative control for the tagging experiment and test them in parallel so you have a good measure of ICC assay specificity. We generally include Sox2-V5 tagging as a positive control in our knock-in experiments as it works quite efficiently and serves as a good control for both the activity of CRISPR reagents and ICC antibody specificity.
- Derive clonal lines by single-cell deposition in 96-well plates: Characterize clonal lines by ICC and PCR genotyping (Sanger sequencing); use primers flanking the tag sequence and expect a size-shift in the PCR product for knock-in lines.
Troubleshooting low knock-in efficiency with the csRNP method
The success of knock-in experiments depends on many factors. Some of these include:
- Most importantly, the crRNA should cut near the insertion site (within ~8-15 bp from the stop codon for C-terminus tagging)
- GC content of the crRNA (aim for 40-60% GC content)
- crRNA activity
- Local chromatin structure (although we could successfully knock-in non-expressed genes with similar efficiency, Dewari et al. 2018).
Using the first crRNA we designed for ~250 genes, we have had about 30% tagging success, although many of these genes were expressed at really low levels so actual knock-in success was ~%50. If you get low knock-in efficiency (<2% positive by ICC for the tag) or do not see any cells positive for tag insertion, you should test a new crRNA and/or try out an alternative tag (3xFLAG, Myc, V5, or HA). Tag-specific PCR genotyping can tell if HDR completely failed. If C-terminus tagging potentially disrupts protein function, an N-terminus tagging approach should be pursued, however, clonal lines must be cautiously analyzed for any damage (indels) to the promoter region of the non-HDR allele. For N-terminal tagging, specifically searching for error-free knock-in at both alleles may be warranted (20-30% of all tagged clones were bi-allelic in our hands).
Take-home message
In our hands, delivering Cas9 protein in complex with custom synthetic cr:tracrRNAs (csRNP) is the most efficient and convenient choice for epitope tagging in diverse stem cell types. Hundreds of genes can be epitope-tagged in a couple of weeks by combining our “TAG-IN” design tool with the csRNP method. We anticipate that our method will open up immense possibilities to delineate the function of large protein cohorts in diverse mammalian cell types. A detailed step by step protocol for the csRNP method is described in our recent article (Dewari et al. 2018), however, if you have any questions, do not hesitate to contact me at Pooran.Dewari@ed.ac.uk. Keep pipetting and happy genome editing!
 Pooran Dewari is a postdoc in Steve Pollard’s lab at the MRC Centre for Regenerative Medicine (CRM), Edinburgh. His research interests include optimizing CRISPR/Cas9 genome-editing methods and applying them to study brain tumour biology. When not pipetting, he can be seen daydreaming about sunny days in the UK.
Pooran Dewari is a postdoc in Steve Pollard’s lab at the MRC Centre for Regenerative Medicine (CRM), Edinburgh. His research interests include optimizing CRISPR/Cas9 genome-editing methods and applying them to study brain tumour biology. When not pipetting, he can be seen daydreaming about sunny days in the UK.
References
1. Bressan RB, Dewari PS, Kalantzaki M, Gangoso E, Matjusaitis M, Garcia-Diaz C, Blin C, Grant V, Bulstrode H, Gogolok S et al. 2017. Efficient CRISPR/Cas9-assisted gene targeting enables rapid and precise genetic manipulation of mammalian neural stem cells. Development 144: 635-648. PubMed PMID: 28096221. PubMed Central PMCID: PMC5312033.
2. Dewari PS, Southgate B, Mccarten K, Monogarov G, O'Duibhir E, Quinn N, Tyrer A, Leitner MC, Plumb C, Kalantzaki M et al. 2018. An efficient and scalable pipeline for epitope tagging in mammalian stem cells using Cas9 ribonucleoprotein. Elife 7. PubMed PMID: 29638216. PubMed Central PMCID: PMC5947990.
3. Jarvik JW, Telmer CA. 1998. Epitope tagging. Annu Rev Genet 32: 601-618. PubMed PMID: 9928493.
4. Liang X, Potter J, Kumar S, Ravinder N, Chesnut JD. 2017. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol 241: 136-146. PubMed PMID: 27845164.
5. Clarke, Ryan, et al. "Enhanced bacterial immunity and mammalian genome editing via RNA polymerase-mediated dislodging of Cas9 from double strand DNA breaks." bioRxiv(2018): 300962. PubMed PMID: 29979968.
Additional Resources on the Addgene Blog
- Plasmids for endogenous gene tagging in human cells
- Plasmids 101: Protein Tags
- Fluorescent tagging of endogenous genes with SapTrap
Resources on Addgene.org
- Browse CRISPR plasmids
- Check out our CRISPR guide pages
- Find ready-to-use Cas9 lentivirus
Topics: CRISPR, Other CRISPR Tools





Leave a Comment