This post was contributed by Jacob Lazarus, a postdoctoral researcher at Harvard.
There’s an astounding number of ways to create chromosomal mutations in bacteria, so many that it may be difficult to decide which path to take. A quick and easy way to introduce a mutation in the chromosome is to disrupt expression of a gene with an antibiotic resistance cassette. This leaves a “scar” in the chromosome, sometimes interfering with expression of surrounding genes. However, there are ways to create scarless mutations, ones that don’t leave any undesired scars in the chromosome.
These methods roughly fall into two categories: those that rely on exogenous enzymes and those that use endogenous homologous recombination.Modifying bacterial genomes using exogenous enzymes: Lambda red and CRISPR
Developed over the past 20 years, Lambda Red recombineering, which uses enzymes domesticated from the Lambda bacteriophage, is arguably the most widely and productively used (Thomason et al., 2014). In recent years, CRISPR-Cas9 technology has also aided in the creation of scarless mutations, both in stand-alone systems (Jiang et al., 2013), and those which increase the efficiency of Lambda red (Pyne et al., 2015; Reisch and Prather, 2015).
Both of these systems are powerful, but they rely on exogenous enzymes, require multiple transformation and plasmid curing steps, and can generate off-target mutations.
Modifying bacterial genomes using counter-selectable markers and homologous recombination
As an alternative, endogenous homologous recombination enzymes can also accomplish the desired genomic modification. This technique is frequently called allelic exchange. Here, a mutated allele introduced from a conjugative plasmid replaces the chromosomal copy. This is a versatile technique, and is less prone to off-target mutations. Ultimately, successful allelic exchange requires two sequential homologous recombination events, but because these events are rare, each requires selection.
These two steps are (Figure 1):
- The initial plasmid integration step (the so-called “single crossover”) which results in a merodiploid. This can be accomplished by selecting for an antibiotic resistance marker from the vector (abR, Figure 1).
- The resolution step (“double crossover”) which leads to the desired mutation and excision of the vector backbone. This step requires “counter-selection,” meaning that the selection is for the loss of a specific phenotype (using sacB, Figure 1, further discussed below).
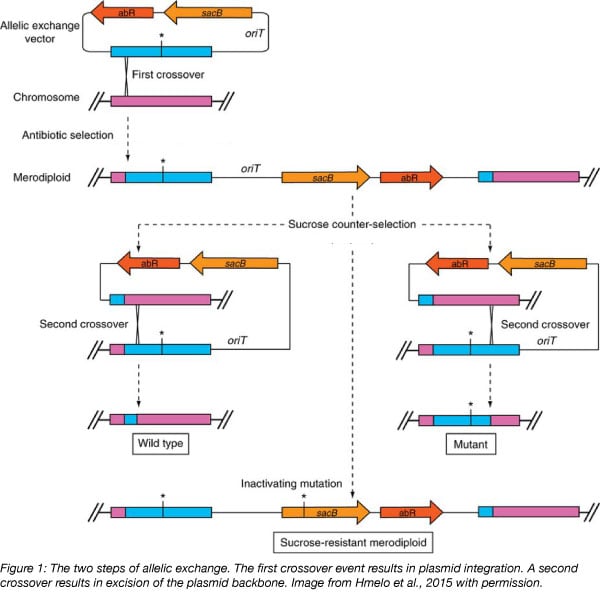
Counter-selectable marker: sacB
What are the counter-selectable markers used to identify double crossovers? The most commonly used counter-selection system relies on the sacB gene product of B. subtilis (Hmelo et al., 2015). The sacB gene product is a sucrase that converts sucrose into a toxic metabolite. Cells that have undergone a double crossover will not contain sacB, allowing them to grow on sucrose. Cells that have only gone through the single cross over still contain sacB and will not grow on sucrose.
One popular counterselection vector is the pDS132 sacB-based allelic exchange vector. This vector contains sacB, an chloramphenicol resistance cassette, and the R6K origin of replication to allow replication only in hosts carrying the bacteriophage lambda pir protein. This allows conjugation between a pir-containing donor and the recipient of interest. Because pDS132 cannot be replicated in the recipient, the only antibiotic resistant colonies isolated are those where the allelic exchange vector has undergone single crossover and integrated into the genome. Subsequent selection on sucrose will identify double crossovers.
This can be an efficient method to generate precise, unmarked mutants, but it frequently requires considerable optimization to ensure strong counter-selection. Anecdotally, there is also a large amount of user-to-user variability, and in some cases, counter-selection escape can occur: single crossover colonies can acquire the ability to grow on sucrose, perhaps by sacB inactivation.
Bacterial toxins for counter-selection
In 2015, the Chen lab harnessed a group of bacterial toxins to facilitate efficient recombineering (Khetrapal et al., 2015). They created vectors for Lambda Red that contained one of eight bacterial toxin genes isolated from E. coli or Psuedomonas aeruginosa. These toxins were a mix of Type VI secreted effector toxins, toxin-antitoxin type toxins, and phage holins. They hypothesized that genes that had evolved specifically for toxicity would allow stronger counter-selection than one merely co-opted toward that aim (i.e. sacB).
Importantly, to prevent activation of toxins before the second crossover step, they tested their expression under both rhamnose- and arabinose-inducible promoters. Tight control of toxin expression is absolutely necessary, because leaky expression of toxin could select for mutations in the toxin itself (leading to counter-selection escape). The rhamnose induction system is well-characterized and quite tight (Giacalone et al., 2006). The Chen lab demonstrated excellent counter-selection which enabled very efficient recombineering.
We were inspired by this work and sought to develop these negative selection markers for allelic exchange.
To do this, we performed the following modifications to pDS132 (Philippe et al., 2004):
- Replaced the sacB cassette with one of three toxins from the Khetrapal paper. We selected the three toxins so that at least one might be useful in a broad array of pathogenic Proteobacteria. We placed the toxin genes under control of a rhamnose-inducible promoter, though Khetrapal et al. also demonstrated that the arabinose promoter worked too.
- Rhamnose induction relies on the transcriptional activator rhaS. To allow manipulation of bacteria without an endogenous rhamnose regulon, we inserted a copy of rhaS with a strong synthetic ribosome binding site (using the online calculator from the Salis group) (Espah Borujeni et al., 2014).
- Replaced the pDS132 MCS with a versatile, synthetic MCS (designed with the help of the DNA tuner program) (Latynski and Valentovich 2014). We routinely clone the purified amplicons upstream and downstream of the desired deletion directly into cut vector with excellent efficiency using a commercial isothermal assembly master mix.
- Introduction of the strong transcriptional terminator BBa_B1002 (from IGEM) upstream of the MCS to reduce transcriptional readthrough into the MCS to facilitate the cloning of toxic products.
- To allow manipulation of naturally chloramphenicol-resistant strains, we have also included versions with gentamicin-resistance (amplified from TP997).
 We validated these vectors, which we call “pTOX” in a broad group of Gram-negative Enterobacteriaceae including in E. coli O157:H7, Shigella flexneri, Enterobacter cloacae, and Serratia marcescens and have created dozens of deletions as well as insertions with this system (Lazarus et al., 2019).
We validated these vectors, which we call “pTOX” in a broad group of Gram-negative Enterobacteriaceae including in E. coli O157:H7, Shigella flexneri, Enterobacter cloacae, and Serratia marcescens and have created dozens of deletions as well as insertions with this system (Lazarus et al., 2019).
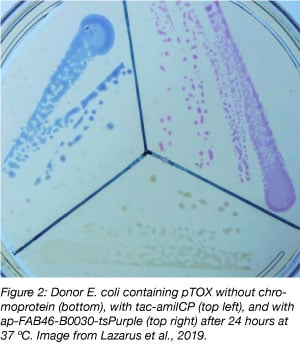
Additionally, to facilitate modification of strains where conjugation may be difficult, we have also included versions of pTOX that encode one of two visible blue or magenta chromoproteins (Figure 2) (Liljeruhm et al., 2018). This system is a nice confirmation of successful single crossover – after about 48 hours, the resulting colonies turn subtly blue or magenta. This is particularly valuable when first optimizing conjugation in strains that don’t conjugate that well. This is because in these circumstances, the conjugation can be so inefficient, that instead of the resulting colonies being true single crossovers, they are just spontaneous antibiotic resistant clones. But if they are blue or magenta, they are real!
Planning an allelic exchange experiment? Find the pTOX plasmids here!
Continued development of new allelic exchange vectors
Allelic exchange is a great technique for the scarless modification of bacterial genomes. It can accomplish large deletions, insertions, and even precise point mutations. I think it will always have a place in every bacterial geneticist’s toolbox.
Depending on the growth rate of the organism of interest, from the day primers arrive, the desired chromosomal modification can be made in 5 days.
Counter-selection with sacB has revolutionized allelic exchange. Even now, only half our lab has switched over to pTOX. However, we feel it has clear advantages, and are hopeful that continued development of allelic exchange vectors will produce an array of great options, and continue to improve the efficiency of this versatile technique.
Many thanks to our guest blogger Jacob Lazarus from Harvard.
 Jake received his MD and a PhD in Cell and Molecular Biology from the University of Pennsylvania, and trained in adult medicine and clinical infectious diseases at the Massachusetts General Hospital where he is now an attending physician. He performs post-doctoral research in the lab of Matthew Waldor at Harvard. Now that he has an efficient allelic exchange system, he is so excited to use it to explore how hospital-associated Gram-negative bacteria become resistant to antibiotics!
Jake received his MD and a PhD in Cell and Molecular Biology from the University of Pennsylvania, and trained in adult medicine and clinical infectious diseases at the Massachusetts General Hospital where he is now an attending physician. He performs post-doctoral research in the lab of Matthew Waldor at Harvard. Now that he has an efficient allelic exchange system, he is so excited to use it to explore how hospital-associated Gram-negative bacteria become resistant to antibiotics!
References
Espah Borujeni, Amin, Anirudh S. Channarasappa, and Howard M. Salis. "Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites." Nucleic acids research42.4 (2013): 2646-2659. PubMed PMID: 24234441. PubMed Central PMCID: PMC3936740.
Giacalone, Matthew J., et al. "Toxic protein expression in Escherichia coli using a rhamnose-based tightly regulated and tunable promoter system." Biotechniques 40.3 (2006): 355-364. PubMed PMID: 16568824.
Hmelo, Laura R., et al. "Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange." Nature protocols 10.11 (2015): 1820. PubMed PMID: 26492139. PubMed Central PMCID: PMC4862005.
Jiang, Wenyan, et al. "RNA-guided editing of bacterial genomes using CRISPR-Cas systems." Nature biotechnology31.3 (2013): 233. PubMed PMID: 23360965. PubMed Central PMCID: PMC3748948.
Khetrapal, Varnica, et al. "A set of powerful negative selection systems for unmodified Enterobacteriaceae." Nucleic acids research 43.13 (2015): e83-e83. PubMed PMID: 25800749. PMCID: PMC4513841.
Latynski US, Valentovich LN. "DNA tuner: a computer program for the construction of polylinker sequences of molecular vectors". Proceedings of the Belarusian State University Series of Physiological, Biochemical and Molecular Biology Sciences. 9.1 (2014) :148–153. [accessed 2019 Feb 28].
Lazarus, Jacob E., et al. "A New Suite of Allelic Exchange Vectors for the Scarless Modification of Proteobacterial Genomes." Applied and environmental microbiology (2019): AEM-00990. PubMed PMID: 31201277.
Liljeruhm, Josefine, et al. "Engineering a palette of eukaryotic chromoproteins for bacterial synthetic biology." Journal of biological engineering 12.1 (2018): 8. PubMed PMID: 29760772. PubMed Central PMCID: PMC5946454.
Philippe, Nadège, et al. "Improvement of pCVD442, a suicide plasmid for gene allele exchange in bacteria." Plasmid 51.3 (2004): 246-255. PubMed PMID: 15109831.
Pyne, Michael E., et al. "Coupling the CRISPR/Cas9 system with lambda red recombineering enables simplified chromosomal gene replacement in Escherichia coli." Appl. Environ. Microbiol. 81.15 (2015): 5103-5114. PubMed PMID: 26002895. PubMed Central PMCID: PMC4495200.
Reisch, Chris R., and Kristala LJ Prather. "The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli." Scientific reports 5 (2015): 15096. PubMed PMID: 28060411.
Thomason, Lynn C., et al. "Recombineering: genetic engineering in bacteria using homologous recombination." Current protocols in molecular biology 106.1 (2014): 1-16. PubMed PMID: 24733238.
Additional resources on the Addgene blog
- Read our Plasmids 101 blog posts
- Find our CRISPR blog posts
- Learn more about Lambda Red recombineering
Resources on Addgene.org
- Find plasmids for allelic exchange
- Read our molecular biology reference
- Watch molecular biology protocol videos
Topics: Molecular Biology Protocols and Tips, Other Plasmid Tools, Plasmids





Leave a Comment