Originally published Mar 3, 2016 and last updated Apr 13, 2021 by Will Arnold.
Although CRISPR systems were first discovered in bacteria, most CRISPR-based genome engineering has taken place in other organisms. In many bacteria, unlike other organisms, CRISPR-induced double stranded breaks are lethal because the non-homologous end-joining (NHEJ) repair pathway is not very robust. In many cases, homology-directed repair does not function effectively either, but scientists have devised means of co-opting phage genetic systems to facilitate homologous recombination in bacteria. These quirks change the way CRISPR-mediated genome engineering functions in bacteria, but have no fear - plasmids from Addgene depositors are making it easier than ever to use CRISPR in E. coli and other bacterial species. Read on to learn about the tools available for bacteria and some of the applications for which they’ve been used.
The beginnings of bacterial CRISPR engineering
One common way of accomplishing bacterial genome engineering is done with recombineering, a technique that utilizes phage recombination machinery to promote homologous recombination of linear DNA fragments. Since recombineering does not contain a selection step for successful modifications, efficiency can be low, especially for larger modifications.
What’s the solution to this inefficiency? Use CRISPR to make it a selectable process! As NHEJ is ineffective in bacteria, CRISPR-induced double stranded breaks (DSB) are lethal. Addgene depositor Luciano Marraffini’s lab took advantage of this lethality to design the first synthetic bacterial CRISPR system in E. coli. The system available from Addgene consists of two plasmids:
- pCas9: carries Cas9 and chloramphenicol resistance
- pCRISPR: carries a spacer targeting the gene of interest and kanamycin resistance
E. coli carrying the phage recombineering machinery are first electroporated with pCas9. Then, pCRISPR is introduced along with an oligonucleotide repair template. Through recombineering, the locus of interest is modified to match the repair template, and the locus cannot be recognized by the spacer-derived crRNA. However, if recombineering is unsuccessful and the wild-type sequence persists, Cas9 will cleave the gene of interest, inducing a lethal DSB.
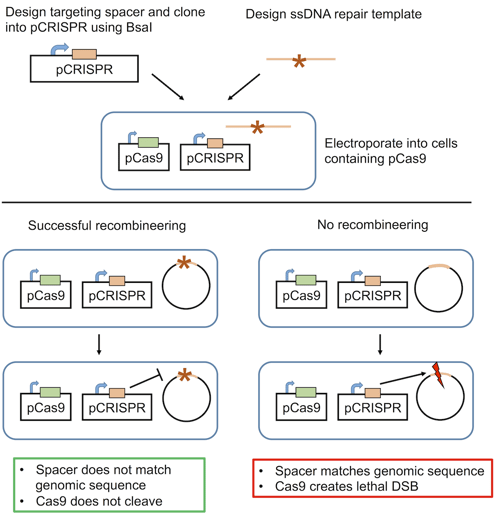
This system is distinct from those used in eukaryotes in that CRISPR isn’t the primary editing force. In contrast, in E. coli, CRISPR is primarily a means of selection that targets cells in which homologous recombination has not occurred. This powerful negative selection system ensures high editing efficiency; the only non-edited cells to survive have inactivating mutations in the Cas9 or spacer sequence, and these rare events are easily detectable using PCR. The system also functions in S. pneumoniae and can be used to generate multiple mutations simultaneously (Jiang et al. 2013).
Genome editing tools for bacteria
CRMAGE system combines CRISPR and recombineering-based MAGE
The Nielsen lab’s CRMAGE system is a fast, multiplexable method that combines CRISPR and recombineering-based MAGE (Multiplex Automated Genome Engineering) technology (Ronda et al., 2016). pMA7CR_2.0 expresses lambda Red and Cas9, which are separately inducible by L-arabinose and anhydrotetracycline (aTet), respectively. pMAZ-SK contains an aTet-inducible gRNA and a backbone-targeting gRNA cassette for plasmid curing through "self-destruction" after induction with L-rhamnose and aTet. CRMAGE is much more efficient than traditional recombineering, with 96-99% efficiency for point mutations and 66% efficiency for small insertions. Multiplexing of two targets simultaneously is possible with efficiency >70%. CRMAGE is an incredibly fast protocol, with only 5 hours incubation time needed for a single round of editing, and a subsequent curing protocol that requires only 2-3 hours incubation.
E. coli and T. citrea scarless editing plasmids
Sheng Yang’s lab describes a two-plasmid system that combines recombineering with CRISPR to create a system for scarless, iterative genome engineering (Jiang et al., 2015). pCas contains Cas9 and the phage recombination gene lambda Red. pTargetF contains the specific gRNA(s), and the repair template is supplied as a dsDNA fragment. Each round of editing takes two days, and the pTargetF and pCas plasmids can be subsequently cured from the bacteria. Although developed in E. coli, the system was used successfully in Tatumella citrea, another species of Enterobacteriaceae, without the need for modification. This finding suggests that the system may be functional in most Enterobacteriaceae.
An update to this system, called pEcCas/pEcgRNA, allows it to be used in more E. coli strains including the popular BL21(DE3) (Li et al., 2021). This pEcCas/pEcgRNA system works similar to the original pCas/pTargetF but has many other advantages including shorter plasmid curing time.
pCRISPomyces for editing in Streptomyces
Streptomyces bacteria produce a wide variety of bioactive natural products. To easily explore and engineer pathways within this genus, Huimin Zhao’s lab created two “pCRISPomyces” systems for use in Streptomyces (Cobb et al., 2015). pCRISPomyces-1 includes Cas9, a tracrRNA, and a CRISPR array, while pCRISPomyces-2 contains Cas9 and a gRNA cassette. The simpler system of pCRISPomyces-2 displays a higher editing efficiency, perhaps due to its condensed design. For both systems, custom spacers/gRNAs are easily inserted using BbsI and Golden Gate Assembly. Either plasmid can be also linearized with XbaI to insert extra elements, like a repair template, using Gibson Assembly or restriction enzyme cloning. Streptomyces bacteria are more recombinogenic than E. coli, so this system functions more similarly to CRISPR/Cas9 systems adapted for eukaryotes in that Cas9-mediated cleavage induces HDR to directly modify a given gene.
CRISPR-tranposons
By combining CRISPR editing and transposons, the Sternberg and Zhang labs developed CRISPR-transposons. The Sternberg lab's system called INTEGRATE (Insertion of Transposable Elements by Guide RNA-Assisted Targeting) and the Zhang lab's system called CAST (CRISPR-associated Transposase) both enable gRNA-directed transposition. INTEGRATE consists of four major components including (1) a CRISPR RNA, (2) four proteins forming the QCascade DNA-targeting module with the crRNA, (3) three transposase proteins, and (4) the donor DNA. By using a multi-spacer CRISPR array, CRISPR-transposons can be multiplexed.
Transcriptional repression (CRISPRi) in bacteria
As RNA interference does not function in bacteria, most efforts to regulate gene expression were limited to inducible promoters or direct gene knockout. In contrast, CRISPR offers a much more user-friendly way to modulate gene expression. The Marraffini lab and Qi lab developed early systems for E. coli; while the Marraffini lab used a native minimal CRISPR array (Bikard et al., 2013), the Qi lab employed a gRNA-based design more familiar to those using CRISPR in eukaryotes (Qi et al., 2013). As in other systems, catalytically dead (dCas9) targeted to a promoter or gene body can repress transcription by physically blocking the elongation complex from binding the DNA or extending the transcript. Different Cas orthologues and subtypes function somewhat differently and can be more effective targeting either the promoter or the coding sequence and the template or non-template strand, among other parameters. .
Stanley Qi lab's CRISPRi plasmids
Stanley Qi’s lab demonstrated that a minimal, regulatable CRISPR system effectively silenced transcription of one or more genes in E. coli. and in a mammalian cell line (Qi et al., 2013). Two important features of this early system are its regulatable nature and its ability to multiplex repression. Using a Tet responsive promoter to drive expression of a catalytically dead Cas9 enzyme allows reversible repression based on the presence of the inducer, in this case anhydro-tetracycline (aTc). By cloning in two tandem copies of the gRNA cassette, one can reliably knockdown expression of multiple target transcripts simultaneously.
Marraffini lab's CRISPRi/a plasmids
In addition to the early work of the Marraffini lab pioneering genome editing using CRISPR in bacteria, they also led early efforts creating tools for CRISPRi and CRISPRa (Bikard et al., 2013). By targeting either a catalytically dead Cas9 (dCas9) alone or a dCas9 fused to the omega subunit of RNA polymerase, the lab successfully repressed or activated transcription of select genes in E. coli. Furthermore, to demonstrate wider applicability they demonstrated success of Cas9 mediated repression in the Gram-positive bacterium, Streptococcus pneumoniae.
Robert Husson lab's M. tuberculosis CRISPRi plasmids
A group from the Husson lab used CRISPRi to study essential genes in M. tuberculosis. dCas9 and a gRNA are expressed on pRH2502 and pRH2521, respectively. Both dCas9 and the gRNA are Tet-inducible, and a gRNA can be cloned easily into pRH2521 using BbsI. This system results in 80-90% RNA knockdown across multiple gRNAs per gene (Singh et al. 2016).
Sarah Fortune's lab M. tuberculosis CRISPRi plasmids
Noting that the more common spCas9 system has generally low efficiencies or can be proteotoxic when used in Mycobacterial species, Sarah Fortune’s lab screened 11 other Cas9 orthologs to identify a more efficient, well tolerated, Cas enzyme. They identified the Cas9 orthologue from Streptococcus thermophilus, Cas9sth1 as a robust and highly active enzyme for transcriptional silencing Mycobacterium tuberculosis and Mycobacterium smegmatis. In addition to their identification of a new CRISPRi tool, they also optimized the system by modulating the PAM to further tune efficiency of knockdown and developed new, more tightly controlled, Tet responsive promoters to avoid leaky activation of the system.
Mobile CRISPRi for Gram-negative and Gram-positive bacteria
Jason Peters, Oren Rosenberg, and Carol Gross used two different genetic systems, targeting either Gram-negative bacteria or Gram-positive firmicutes, to bring CRISPRi to a much wider swath of bacterial species (Peters et al., 2019). The systems differ on how exactly the CRISPRi locus is introduced (Tn7 in Gram-positives and ICEbs1 in Gram-negatives) but share the common CRISPR-dCas9 system for targeted repression of gene expression. They successfully knock down expression of essential and non essential genes in a wide variety of bacteria including in pooled, genome-wide, formats. Interested in knowing more? Check out our Mobile CRISPRi blog post.
CRISPR activation (CRISPRa)
While the activation of transcription is a more challenging problem than repression, scientists have deposited several plasmid tools that can be used in this way. As described in the above CRISPRi section, the Marraffini Lab published a robust tool for CRISPRi and CRISPRa using RNAP-Omega-dCas9 fusions. Here’s some others.
SoxS based CRISPRa
In the search for a CRISPR activation method in E. coli, Jesse Zalatan’s lab used an approach to identify known proteins involved in the activation of transcription including transcription factors, phage proteins, and RNAP components (Dong et al., 2018). They found a transcription factor called SoxS for CRISPRa. Adding complexity, they found that they could also include a CRISPRi system for multiplexed modulation.
CRISPRa from sigma-54 promoters
Noting that previous work in bacterial CRISPRa had focused on sigma-70 dependent promoters, Baojun Wang’s lab developed a system that can activate sigma-54 dependent promoters (Liu et al., 2019). While sigma-70 promoters are ubiquitous, they are not universal, particularly when it comes to non-model bacteria. The authors were able to engineer a dCas9 variant fused to bacterial enhancer binding protein required for the activation of sigma-54 type promoters. The system was functional in activating promoters in Pseudomonas syringae and Klebsiella oxytoca.
Microbiome engineering
Scientists have begun to realize the potential CRISPR technology can have on the human microbiome. You can consider the microbiome as a collection of organisms, their genes, and metabolic processes, occupying the gut. Many human diseases have connections to either specific organisms or organism/gene interactions with the host.
This scenario allows for several potential CRISPR applications. First, CRISPR can be used to target locations in the genomes of pathogenic or undesirable bacteria. This system can be delivered a number of ways but success has been observed with Phagemids (Selle et al., 2020, Citorik et al., 2015, Bikard et al., 2014). When the CRISPR system targets these genomic locations, and introduces double strand breaks, the bacteria are unable to resolve them, and the cells die.
This could be applied to diseases where the presence of just a single organism, such as Clostridium difficile, could be targeted and preferentially depleted from gut. While still far from the clinic, CRISPR-Cas3 systems have been used successfully in mice for just such a purpose (Selle et al., 2020).
Targeting a subset of bacteria in a population
Work from the Lu lab found early success using CRISPR-Cas to target subsets of bacteria in population based on the presence of specific target sequences (Citorik et al., 2014). These could be genes, specific polymorphisms, including antibiotic resistance genes. This approach successfully targeted specific organisms when using bacteriophages or mobile plasmids in a bacterial strain introduced to the population as methods of introduction.
Engineering the bee gut microbiome
Beyond the human gut, the Barrick lab has deposited plasmids that can be used to engineer the bee gut microbiome. They created a modular system using a broad host range plasmid that can be maintained in a number of bacteria native to the honey bee and bumble bee gut microbiomes titling it the Bee microbiome toolkit (Leonard et al., 2018).
Base Editing
Among the newest approaches that leverage the remarkable properties of CRISPR-Cas systems is base editing. This process is generally accomplished by fusing an enzyme capable of converting one base to another to dCas9. The typical gRNA targeting approach is then used to direct the complex to a specific locus. This approach provides a more granular level of control where individual bases can be modified.
Cytidine deaminase PmCDA1
Akihiko Kondo’s lab identified a cytidine deaminase, PmCDA1, that could be fused to dCas9 and efficiently introduce cytosine-to-thymine substitutions in target sequences (Banno et al., 2018). These substitutions were limited to approximately a five-base pair window of the target sequence which could be adjusted by changing the sgRNA length. The system could be further refined by the addition of a uracil DNA glycosylase inhibitor and degradation tag on the fusion protein to limit the activity of the system as a whole.
Cytidine deaminase for Pseudomonas species
Expanding this tool to other organisms, the Ji lab brought genome and base editing to Pseudomonas species (Chen et al., 2018). Using the cytidine deaminase, APOBEC1 and a nickase version of Cas9 they were effectively able to introduce point mutations in P. aeruginosa, Pseudomonas putida, Pseudomonas fluorescens, and Pseudomonas syringae. These vectors contain BsaI sites for easy cloning of a gRNA of interest.
Cytidine and adenine base editing in Streptomyces
Some of the most industrially and clinically relevant bacterial species reside in the genus Streptomyces, yet genome engineering remains challenging for most organisms in the genus. Work from the Sun lab demonstrates the utility of cytidine and adenine base editors that enable C-to-T and A-to-G base substitutions respectively. Substitutions seem most efficient when using a nickase version of Cas9, but were also successful using a dCas9 variant as well. Furthermore, they provide a high-fidelity (HF) variant of the BE3 that further improves editing rates.

References
Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, Marraffini LA (2014) Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol 32:1146–1150 . https://doi.org/10.1038/nbt.3043
Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA (2013) Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Research 41:7429–7437 . https://doi.org/10.1093/nar/gkt520
Citorik RJ, Mimee M, Lu TK (2014) Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol 32:1141–1145 . https://doi.org/10.1038/nbt.3011
Cobb RE, Wang Y, Zhao H (2014) High-Efficiency Multiplex Genome Editing of Streptomyces Species Using an Engineered CRISPR/Cas System. ACS Synth Biol 4:723–728 . https://doi.org/10.1021/sb500351f
Dong C, Fontana J, Patel A, Carothers JM, Zalatan JG (2018) Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat Commun 9: . https://doi.org/10.1038/s41467-018-04901-6
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA (2013) RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31:233–239 . https://doi.org/10.1038/nbt.2508
Leonard SP, Perutka J, Powell JE, Geng P, Richhart DD, Byrom M, Kar S, Davies BW, Ellington AD, Moran NA, Barrick JE (2018) Genetic Engineering of Bee Gut Microbiome Bacteria with a Toolkit for Modular Assembly of Broad-Host-Range Plasmids. ACS Synth Biol 7:1279–1290 . https://doi.org/10.1021/acssynbio.7b00399
Li Q, Sun B, Chen J, Zhang Y, Jiang Y, Yang S (2021) A modified pCas/pTargetF system for CRISPR-Cas9-assisted genome editing in Escherichia coli. Acta Biochimica et Biophysica Sinica. https://doi.org/10.1093/abbs/gmab036
Liu Y, Wan X, Wang B (2019) Engineered CRISPRa enables programmable eukaryote-like gene activation in bacteria. Nat Commun 10: . https://doi.org/10.1038/s41467-019-11479-0
Peters JM, Koo B-M, Patino R, Heussler GE, Hearne CC, Qu J, Inclan YF, Hawkins JS, Lu CHS, Silvis MR, Harden MM, Osadnik H, Peters JE, Engel JN, Dutton RJ, Grossman AD, Gross CA, Rosenberg OS (2019) Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat Microbiol 4:244–250 . https://doi.org/10.1038/s41564-018-0327-z
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA (2013) Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 152:1173–1183 . https://doi.org/10.1016/j.cell.2013.02.022
Ronda C, Pedersen LE, Sommer MOA, Nielsen AT (2016) CRMAGE: CRISPR Optimized MAGE Recombineering. Sci Rep 6: . https://doi.org/10.1038/srep19452
Selle K, Fletcher JR, Tuson H, Schmitt DS, McMillan L, Vridhambal GS, Rivera AJ, Montgomery SA, Fortier L-C, Barrangou R, Theriot CM, Ousterout DG (2020) In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. mBio 11: . https://doi.org/10.1128/mbio.00019-20
Singh AK, Carette X, Potluri L-P, Sharp JD, Xu R, Prisic S, Husson RN (2016) Investigating essential gene function inMycobacterium tuberculosisusing an efficient CRISPR interference system. Nucleic Acids Res 44:e143–e143 . https://doi.org/10.1093/nar/gkw625
Resources on the Addgene Blog
- Check out our CRISPR Featured Topic Page
- Learn more about Cpf1
- Find out about CRISPR Multiplexing
Resources on Addgene.org
- Browse our CRISPR Guide Pages
- Find Bacterial CRISPR Plasmids
Topics: CRISPR, CRISPR 101, CRISPR Expression Systems and Delivery Methods





Leave a Comment