Originally published Apr 13, 2021 and last updated Aug 12, 2021.
This post was contributed by guest bloggers, Yiwen Zhang, a Ph.D. candidate in the Yang Lab, Qi Li, a lecturer at Sichuan Normal University, Jieze Zhang, an intern in the Yang Lab, and Sheng Yang, a professor in synthetic biology from the Chinese Academy of Sciences.
In the Yang lab, we’re continually optimizing the use of CRISPR editing systems in bacteria. Our pCas/pTargetF system has been one of the most popular genomic editing systems in E. coli. However, some users pointed out that this system did not function properly in E. coli BL21(DE3) as transformants failed to grow after the transformation of pTargetF into the BL21(DE3)/pCas strain. In response, we modified the pCas/pTargetF system and updated it into a pEcCas/pEcgRNA system. This system works well for engineering single genome edits but the double strand breaks it introduces makes it difficult to perform two or more integrations on the genome. Most recently, we’ve developed a new system “Multicopy chromosomal integration using CRISPR-associated transposases” or MUCICAT.
Here, we’ll describe each of these CRISPR systems.
The original pCas/pTargetF system
Our original pCas/pTargetF system consists of one plasmid that expresses the S. pyogenes Cas9, a temperature-sensitive replicon for self-curing, an arabinose inducible lambda-red gene, and an IPTG-inducible sgRNA sequence targeting the pMB1 replicon on pTargetF (Jiang et al., 2015). In addition to the pMB1 replicon, pTargetF contains the sgRNA targeting the genomic site of interest. The donor template is supplied separately.
To use this system, you’d first introduce your sgRNA sequence into the pTargetF construct and then introduce both plasmids into the cell. The edit in the chromosome can now take place. After editing, pTargetF can be cured from the cell by inducing expression of the sgRNA that targets the pMB1 replicon, resulting in cutting of the plasmid.
At this point, subsequent introductions of pTargetF containing another sgRNA targeting another site of interest can be used to make other edits. Once you have all the edits you want, cure pCas9 by growing the cells at 37 °C.

The new pEcCas/pEcgRNA system
We speculated that the gRNA on pCas that is specific to the pMB1 replicon of pTargetF may exhibit a more serious leaky expression in BL21(DE3), which we presume caused pTargetF to be cut by Cas9. This would result in the lack of recovered transformants. Our speculation motivated us to update the pCas/pTargetF system.
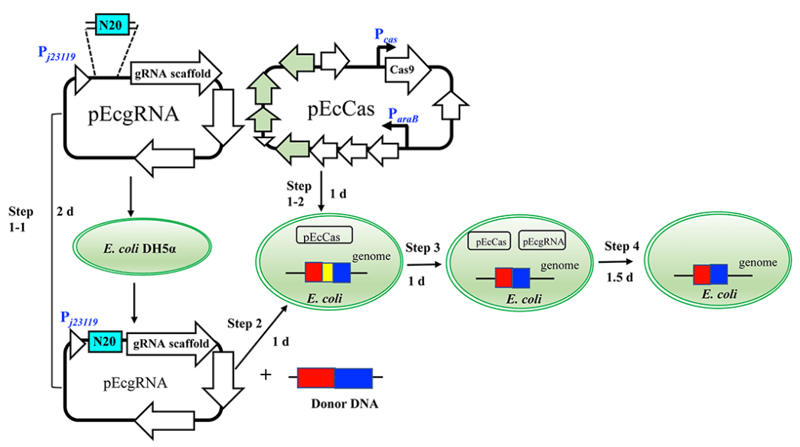 |
| Figure 1: The editing process begins with the addition of the sgRNA sequence in pEcgRNA. Next both plasmids are transformed in E. coli with the donor DNA for editing to take place. Then plasmids are cured from the cell. Steps 1-1 and 1-2 can be done simultaneously. Image from Li et al., 2021. |
The updates involved the following changes. In pCas9, we 1) replaced the promoter of gRNA-pMB1 with PrhaB, 2) changed the replicon of pCas into a non-temperature-sensitive replicon, and 3) added the sacB gene for counterselection to yield pEcCas. In pTargetF, we replaced the original 20 bp specific sequences on pTargetF with the ccdB gene containing BsaI restriction enzyme sites at its ends to generate pEcgRNA. The ccdB serves as a counterselection marker and will be replaced by the sgRNA targeting the genomic site of interest.
We compared the pCas/pTargetF system with the updated pEcCas/pEcgRNA system, and confirmed that indeed the gRNA-pMB1 had a slightly higher leaky expression in BL21(DE3) that resulted in a lower efficiency of transformation for pTargetF series plasmids. Fortunately, we were able to successfully delete genes of interest in BL21(DE3) using the newly generated pEcCas/pEcgRNA system.

Advantages of the pEcCas/pEcgRNA system
Compared with the original system, the pEcCas/pEcgRNA is superior in several ways:
- The pEcCas plasmid can be operated at 37 ℃ for faster growth of its E. coli host, and the addition of a sacB gene renders it beneficial for plasmid clearance.
- It is more convenient to update the spacer(20nt) on pEcgRNA plasmid as only two 24 nt oligos are required and the BsaI linearized pEcgRNA backbone can be prepared in large amounts and frozen for future use or directly ligated with the annealed 24 nt oligos to generate new pEcgRNA.
- pEcCas/pEcgRNA can be applied not only in coli K-12 strains, but also in E. coli B and W strains.
- The optimized plasmid curing method was much simpler and took only ~32 hours instead of ~60 hours.
We successfully tested the pEcCas/pEcgRNA system in 4 E. coli K-12 strains, 2 E. coli B strains, 1 E. coli W strain, and 1 Tatumella citrea, with a total of 70 sites edited. We optimized the strategy for plasmid curing, providing users with a complete editing process.
The MUCICAT system
While the above systems are great for experiments that require only single chromosomal integration (ex: genome editing), our MUCICAT system which is based on the CRISPR-associated transposase system, INTEGRATE, is best for multicopy chromosomal integration (ex: for protein expression) (Zhang et al., 2020). Multicopy chromosomal integration for gene expression is an alternative versus plasmid-based expression which can be unstable and lack precise control over copy number.
MUCICAT system contains 4 plasmids: pDonor, pTnsABC, pQCascade and pCutamp.
- pDonor carries a customizable cargo gene in the form of RE-cargo-LE (RE, right end; LE, left end)
- pTnsABC expresses the transposases TnsA, TnsB, and TnsC
- pQCascade expresses the transposase TniQ, the Cas proteins Cas6, Cas7, Cas8, and the crRNA. The spacers of the crRNA can be customized as well, including concatenations into crRNA arrays targeting multiple different sites in E. coli.
- pCutamp cures the plasmids by targeting the ampR promoter in all three plasmids. pCutamp is then removed by sacB counterselection.
The MUCICAT toolkit includes versions of plasmids with different induction systems, such as PT7, Prha, Ptet, and Para. The pDonor plasmid pRE57-Ter with multiple terminators can serve to interrupt genes (or gene clusters) in metabolic engineering.
So far, we’ve used the MUCICAT system in two ways:
- In our initial paper, we used MUCICAT to optimize the gene dosage of glucose dehydrogenase and generated a strain that could achieve glucose dehydrogenase expression that is 2.6 times higher when compared to the industrial strain.
- In our metabolic engineering case study of N-acetylglucosamine biosynthesis, we constructed a strain library with 1-11 copies of integrated genes. We obtained a stable and high-yielding strain which had 6 times the yield of the industrial strain. Not only is MUCICAT better, it is faster. Using MUCICAT only needs 2 newly constructed plasmids (pDonor and pQCascade) with 4 rounds of transformation to complete the N-acetylglucosamine strain library construction in 8 days. To build an equivalent strain library, our pEcCas/pEcgRNA system would need to construct 13 plasmids with 14 rounds of transformation in 30 days.
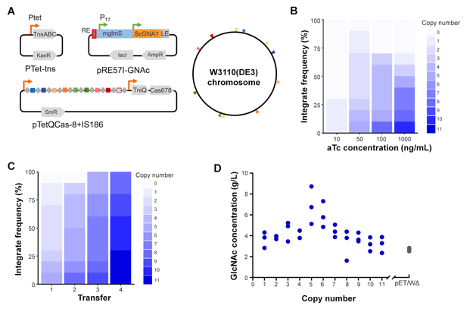 |
|
Figure 2: (A) MUCICAT plasmids for multicopy integration. (B) Anhydrotetracycline (aTc) induces expression transposases leading to increased integration frequency. (C) Increasing rounds of transfer on transposition-inducing agar lead to higher cargo copy numbers. (D) The effect of gene dosage on N-acetylglucosamine concentration. Image from the Yang lab. |
In conclusion, the MUCICAT system has the following advantages:
- Fast and site-specific multicopy chromosomal integration of cargo without using screening markers.
- Capability to construct a strain library with different gene dosages to facilitate swift optimization of strains.
- Multiple induction systems to choose from.
- Ready for metabolic engineering with improved cargo dedicated to gene (cluster) interruption.

Plasmids for each of these systems are available from Addgene. If you’ve given any of these systems a try, we’d love to hear your feedback!
Thank you to our guest bloggers!
 Qi Li obtained her Ph.D. from the Chinese Academy of Sciences (CAS) Center for Excellence in Molecular Plant Sciences in 2019. She joined Sichuan Normal University and became a lecturer in the College of Life Sciences that same year. Her research is focused on the escape mechanism of genome editing in microorganisms which could be applied to optimization of genome editing tools.
Qi Li obtained her Ph.D. from the Chinese Academy of Sciences (CAS) Center for Excellence in Molecular Plant Sciences in 2019. She joined Sichuan Normal University and became a lecturer in the College of Life Sciences that same year. Her research is focused on the escape mechanism of genome editing in microorganisms which could be applied to optimization of genome editing tools.
 Jieze Zhang is an undergraduate student studying biochemistry at the University of Southern California. He is also an intern at the Yang Lab of the CAS Center for Excellence in Molecular Plant Sciences since December 2020. His research focuses on the efficiency of transposon-encoded CRISPR/Cas systems in bacteria, specifically on optimizing strains with low efficiency.
Jieze Zhang is an undergraduate student studying biochemistry at the University of Southern California. He is also an intern at the Yang Lab of the CAS Center for Excellence in Molecular Plant Sciences since December 2020. His research focuses on the efficiency of transposon-encoded CRISPR/Cas systems in bacteria, specifically on optimizing strains with low efficiency.

Sheng Yang obtained his Ph.D. from Shanghai Institute of Biochemistry, Chinese Academy of Sciences. He joined Shanghai Institutes for Biological Sciences in 2000 and became a professor in Synthetic Biology in 2006. The Yang lab's research is focused on developing microbial genome editing tools for faster construction or reprogramming of cells to understand the genetic and biochemical mechanisms behind their distinct phenotypes and to design a new generation of engineered cells for green chemistry and biomedical applications.
 Yiwen Zhang entered the Sheng Yang Lab of the CAS Center for Excellence in Molecular Plant Sciences in September 2019 as a Ph.D. candidate. Her research focuses mainly on the development and application of a multiplex genome editing system based on CRISPR-associated transposition.
Yiwen Zhang entered the Sheng Yang Lab of the CAS Center for Excellence in Molecular Plant Sciences in September 2019 as a Ph.D. candidate. Her research focuses mainly on the development and application of a multiplex genome editing system based on CRISPR-associated transposition.
References and Resources
References:
Jiang Y, Chen B, Duan C, Sun B, Yang J, Yang S (2015) Multigene Editing in the Escherichia coli Genome via the CRISPR-Cas9 System. Appl Environ Microbiol 81:2506–2514 . https://doi.org/10.1128/aem.04023-14
Li Q, Sun B, Chen J, Zhang Y, Jiang Y, Yang S (2021) A modified pCas/pTargetF system for CRISPR-Cas9-assisted genome editing in Escherichia coli. Acta Biochimica et Biophysica Sinica. https://doi.org/10.1093/abbs/gmab036
Additional resources on the Addgene blog:
- Browse CRISPR articles from our CRISPR topic overview
- Find other CRISPR methods for bacteria
- Find all articles about microbiology
Resources on Addgene.org:
- Visit the CRISPR Guide
- Get CRISPR plasmids from Addgene
Topics: CRISPR, Microbiology, CRISPR Expression Systems and Delivery Methods





Leave a Comment