If you are interested in using lentiviral vectors to introduce your favourite gene into your favourite cell line or into primary cells, this blog article will give you some tips to plan your experiment using the lentiviral vector system.
Viral vectors have been increasingly popular in fundamental and applied research since their first use in the early 90’s to genetically modify primary cells. Amongst the different vectors used, lentiviral vector constructs have proven very useful due to their ability to infect both dividing and non-dividing cells, including stem cells. These properties make lentiviral vectors fantastic options for delivering shRNA, CRISPR/Cas9 components, and fluorescent sensors.

Retroviral vector background
Until the development of viral vectors, gene transfer into mammalian cells often relied on chemical (CaCl2, cationic liposome) or electrical (electric current forming membrane pores) transfection. These techniques are still used and efficiently transfer plasmid DNA efficiently into dividing immortalized cells lines but reach their limits when it comes to transferring DNA into primary cells or non-dividing cells. To target these un-transfectable cells, labs use viral vectors and particularly retroviral vectors because they target a broader range of cells and their integration into the genome is more efficient.
The first retroviral vectors used were derived from the Moloney Murine leukemia virus (MoMLV) and consisted of two plasmids: the packaging plasmid containing all the structural genes necessary for the production of a viral particle (Gag, Pol, Tat, Rev and Env) and the transfer plasmid encoding the insert of interest as well as sequences necessary to encapsulate that insert into a viral particle (the packaging signal Psi). Because the packaging plasmids lacked the psi signal, the genes encoding the structural proteins could not be integrated into the new viral particles and the virus particles produced could not replicate. However, Mother Nature found a way to re-create a functional virus. Indeed, by testing several vectors and packaging cell line combinations, Miller et al. showed that infectious wild-type viruses were produced in high-titer retroviral stocks generated by the transfer of a retrovirus into the packaging cell line PA12 (containing the psi deletion). A double crossover event between the vector plasmid and the packaging plasmid caused the Psi sequence to be placed on the packaging plasmid forming a vector that could create replication competent virus. Even though this system was efficient at transferring the gene of interest to the appropriate cells, it was obvious that its safety was a disadvantage particularly for clinical development. In addition, MoMLV is inefficient at infecting non-dividing cells and so researchers set out to create a better, safer system.
Unlike the MoMLV, lentiviruses, a separate genus of the Retroviridae family including human immunodeficiency virus type-1 (HIV-1), can infect both dividing and non-dividing cells. The ability to infect non-dividing cells is not restricted to in vitro cell culture as lentivirus-derived vectors are capable of transducing certain quiescent or terminally differentiated cells such as macrophages and microglia. This property makes the lentivirus an attractive choice for a gene transfer vector.
Lentiviral vectors
Although retroviruses are still in use, considerable efforts have been devoted to develop efficient and safe HIV-1-derived lentiviral vectors due to their ability to infect non-dividing cells. Thus three different generations of lentiviral vectors have been established so far, with safety increasing in each generation.
- The 1st generation of lentiviral vectors consisted of three plasmids encoding 1) the lentiviral vector genome which was composed of the wild-type 5’ and 3’ LTRs, the ψ sequence, a part of the env gene containing the rev response element (RRE), an internal promoter, and the desired gene (transfer vector plasmid), 2) the HIV-1 genome containing all viral genes with the exception of the env gene (packaging plasmid), and 3) the vesicular stomatitis virus G protein (VSV-G) that improves the stability and broadens the cellular tropism of the viral particles produced. However, in this vector production system, there is still a potential risk for the generation of replication competent lentiviruses (RCL) especially if you are working on HIV positive human cell from the clinic.
- The 2nd generation is similar to the first one with the exception that the HIV accessory proteins that are not essential for the production of the lentiviral particle have been removed. This generation is safer than the first one and can be used routinely in a research laboratory. The risk of generating RCL is low but extra care has to be taken especially if you are working with proto-oncogenes or with human samples that have not been tested for HIV. If not using proper technique, there’s always a small chance that a researcher could infect him or herself with the oncogenic virus, or that the accessory factors in HIV positive cells could make the virus replication competent.
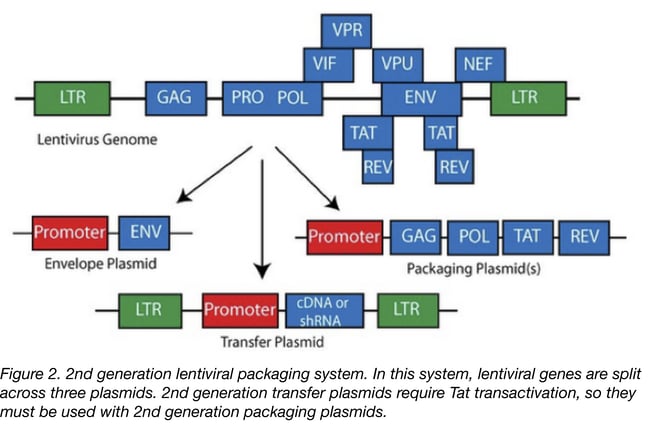
 The 3rd generation of lentiviral vectors was developed for safe use in a clinical setting. In this generation, the HIV Tat gene (previously used to drive expression from the LTRs) has been removed, Rev (which facilitates nuclear export) is expressed from a separate plasmid, and the promoter of the 5’LTR has been deleted to reduce its activity. A CMV or an EF1α promoter has been inserted in this LTR so you don’t need Tat to transcribe the viral genome in producing cells. Currently, the third-generation lentiviral vector system offers the best safety profile in terms of RCL generation because this vector requires only three HIV-1 genes (gag, pol, and rev) for production. Third generation lentiviral vectors can be used for research and for clinical purposes, however improving the safety of these vectors is still an active area of research due to the possibility that mutation or recombination with human retroviruses could lead to RCL.
The 3rd generation of lentiviral vectors was developed for safe use in a clinical setting. In this generation, the HIV Tat gene (previously used to drive expression from the LTRs) has been removed, Rev (which facilitates nuclear export) is expressed from a separate plasmid, and the promoter of the 5’LTR has been deleted to reduce its activity. A CMV or an EF1α promoter has been inserted in this LTR so you don’t need Tat to transcribe the viral genome in producing cells. Currently, the third-generation lentiviral vector system offers the best safety profile in terms of RCL generation because this vector requires only three HIV-1 genes (gag, pol, and rev) for production. Third generation lentiviral vectors can be used for research and for clinical purposes, however improving the safety of these vectors is still an active area of research due to the possibility that mutation or recombination with human retroviruses could lead to RCL.
Regarding transduction efficacy, no differences between the 2nd and the 3rd generation of vectors have been shown. However, with the 3rd generation you get lower viral titer than with the 2nd generation. Knowing this you can scale up your production set-up and purify and concentrate your lentiviral particles to get them to the optimum concentration to infect your cells of interest.
| Advantages | Disadvantages |
| Can carry large transgenes (>8 kb) | Potential for generation of replication-competent lentivirus (RCL) |
| Efficient gene transfer | Potential for insertional mutagenesis: Replication-incompetent lentiviruses with human tropism can still infect the researcher and insert into her/his genome - this is a biosafety risk |
| Infects dividing and non-dividing cells | |
| No immunogenic proteins generated | |
| Stable integration into host genome and stable expression of the transgene |
Transfer plasmids
The transfer plasmid is the vector you clone your gene of interest into and is an important feature for your experiment; it must be chosen carefully. Often researchers want a simple set-up where the gene of interest is expressed together with a marker (Fluorescent protein or selection marker) allowing for selection of the transduced cells. In this case your gene and the marker can be separated by an internal ribosome entry site (IRES) and expressed under the same promoter. Alternatively, your gene and the marker can be placed under the control of two different promoters. Both the single and dual promoter transfer vectors can be used to express your gene of interest either constitutively or inducibly. If your gene of interest is toxic or needs to be expressed a certain point in the cell cycle, a transfer vector with an inducible promoter (see below) should be used.
Inducible lentiviral transfer vectors
The most widely used inducible lentiviral vector system is the tetracycline (Tet)-regulated system. You can choose either the Tet-off system or the Tet-on system. In the Tet-off system a Tet-response element (TRE) is placed upstream of the promoter in your transfer vector. In the absence of tetracycline or a derivative such as Doxycycline (Dox), the Tet-controlled transactivator (tTA, also expressed by your transfer vector) binds to the TRE and activates the expression of your gene. The addition of Dox in the culture will then repress your gene of interest. This system is efficient but you have to be aware that there is often background activity and it requires continuous administration of Dox to repress transgene expression. In contrast, in the Tet-on system, tTA has been modified (and renamed rtTA) to bind TRE only in presence of Dox. In this case, your gene is repressed in steady state and is expressed when Dox is added to the culture. This system is now used as it shows rapid gene expression kinetics as compared to the Tet-off system. One drawback of the Tet system is that it often requires the delivery of two distinct expression vectors to target cells - one containing the gene of interest (pTet-IRES-EGFP, pPRIME-Tet-GFP-FF3) and one with either tTA or rtTA (FUW-M2rtTA). In this kind of approach, a population of cells might express only the tTA or your transgene resulting in low inducibility and potentially making it difficult to interpret your results. This can be a bottleneck for your experiment. Single vector systems containing both your transgene and tTA/rtTA (pINDUCER20, pINDUCER21) have been developed, but these vectors are quite large and you might get lower viral titer when using them.
Regarding biosafety, it is also important to be aware that lentiviral particles are a powerful way to transduce cells - even your own! You should therefore be cautious about inserting potentially harmful genes into a transfer vector. Lentiviral particles expressing proto-oncogenes or oncogenes for instance should be manipulated with care. See the NIH’s biosafety guidelines for more information and always be sure to consult your institution’s Biosafety Committee before beginning any work.
Lentiviral production and transduction
Once you have cloned your gene of interest into lentiviral transfer vector, the next step is to produce the viral particles themselves. For this you will first need to transfect producing cells, usually 293T cells, with your transfer plasmid and your packaging plasmids. For this, you can use the transfection reagent you are most comfortable with. If you don’t have any experience in transfection, CaCl2 transfection is a simple and cheap technique that should work well.
Quickly after transfection (4-8h), cells start releasing viral particles into the culture supernatant and you can generally harvest the supernatant containing viruses 24h after transfection. This supernatant can be stored at 4 °C or can be directly added to your target cells. However in that case you don’t know how many viral particles you put in your culture. As viral production can change from one batch to another. For experimental consistency it’s a good idea to purify and titer your virus. Virus purification can be a tricky process. You have to centrifuge the culture supernatant in order to precipitate the viral particles. Some protocols suggest adding sucrose to the supernatant to create a gradient of density that favours virus precipitation. My tip for this process is to test several protocols that have been described (protocol 1, protocol 2, protocol 3) to find the one that will give you the best production for your system.
See our protocol pages for full Lentivirus Production and Transduction Protocols
Once purified, you must titrate your viral particles to determine their concentration. Three methods are broadly used for virus titration: the p24 ELISA dosing method detects the expression of viral capsid antigen p24, the FACS dosing method correlates viral titre to the number of cells transduced by a GFP encoded virus, and the reverse transcriptase activity assay uses qPCR to quantify viral RNA. All of these methods have been described in a previous Addgene blog
Once you know your viral titre, you can incubate your cells with virus at different multiplicity of infection (MOI=ratio of virus to cells). Again, my tip here is to test several protocols as the set-up depends on the type of cell you would like to infect. Most of the protocols you will find will recommend using a polycation, like polybrene (protocol) or DEAE dextran (protocol), which help the viral particle bind to your target cells. If you are transducing non-adherent cells you might need to centrifuge your cells with the virus in order to aid viral binding. This method, called spinoculation, has been found very effective for T cells transduction for instance.
I hope you will find this information useful for your experiments. Don’t hesitate to check our lentiviral vector guide to get more information and to get access to additional protocols and plasmids.
References
1. Miller, A. DUSTY, and C. A. R. O. L. Buttimore. "Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production." Molecular and Cellular Biology 6.8 (1986): 2895-2902. PubMed PMID: 3785217. PubMed Central PMCID: PMC367857.
2. Naldini, Luigi, et al. "In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector." Science 272.5259 (1996): 263. PubMed PMID: 8602510.
3. Yasutsugu Suzuki and Youichi Suzuki (2011). Gene Regulatable Lentiviral Vector System, Viral Gene Therapy, Dr. Ke Xu (Ed.), ISBN: 978-953-307-539-6, InTech.
4. Walter, W., and U. Stein. "Viral vectors for gene transfer a review of their use in the treatment of human disease." Drugs 60 (2000): 249-71. PubMed PMID: 10983732.
5. Gene Therapy Net Lentiviral Vectors Page
Additional Resources on the Addgene Blog
- 5 Tips for Trouble Shooting Viral Transductions
- Learn Why You Might Want to Use AAV in Your Research
- Lentiviral Vector Uses and Overview
Resources on Addgene.org
- Find Lentiviral Vectors for Your Research
- Order Ready-to-Use Lentivirus
- Browse Our Lentivirus Guide Pages






Leave a Comment