This post was contributed by guest blogger Joachim Goedart, an assistant professor at the Section of Molecular Cytology and van Leeuwenhoek Centre for Advanced Microscopy (University of Amsterdam).
Tagging a protein of interest with a fluorescent protein to study its function is one of the most popular applications of fluorescent proteins. These fusion proteins enable the observation of proteins in living cells and organisms. Both components of the chimera are encoded by DNA. Since researchers can generate almost any DNA sequence in the way that they like, the design and engineering of fusion proteins is relatively straightforward. However, generating a fusion while keeping all of the native properties of the protein of interest can be challenging. In this blog I discuss strategies to generate fusion proteins and highlight some aspects of their design.
 Protein size and shape matters
Protein size and shape matters
The green fluorescent protein (GFP) from Aequorea victoria and its variants are genetically encoded fluorescent probes. One of the limitations is the size of GFP: ~240 amino acids or about 28 kDa. GFP and its homologues have a beta-barrel structure. Although the beta barrel has no strong affinity for other proteins, it may still block the function or activity of the tagged protein because of its size. This type of interference is known as steric hindrance. For instance, GFP may occlude catalytic sites or obstruct binding sites or motifs that are necessary for post-translational modifications. Despite these potential issues, GFP has been used successfully in countless fusion proteins.
Choose monomeric proteins and avoid a sticky situation
Next to size, potential homodimerization and higher order oligomerization of fluorescent proteins is a concern. The propensity of fluorescent proteins to oligomerize is known as “stickiness.” Since the fluorescent tag should operate as an inert light emitting module, any tendency of GFP to homodimerize should be avoided because it will double the size of the protein. In addition, it increases avidity because it doubles the number of binding sites per unit.
There are several ways to measure stickiness. In vitro, ultracentrifugation or gel filtration is the most common analytical method. However, these may not reflect the situation in cells. Therefore, the localization of critical fusions (e.g. tubulin or connexin) has been used as a proxy for dimerization (Shaner et al., 2008 - supplementary figure C). However, these assays are qualitative. The Organized Smooth Endoplasmic Reticulum (OSER) assay (Costantini et al., 2012) measures homodimerization of FPs targeted to the endoplasmid reticulum based on the appearance of OSER whorls. This assay is quantitative and is currently the standard for assessing oligomerization in cells.
In addition to measuring stickiness, several researchers have also collected data on fluorescent protein characteristics such as the brightness, photostability, maturation time, and acid sensitivity of various fluorescent proteins (Botman et al, 2018; Cranfill et al, 2016; Lambert, 2019). These characteristics are important, but have less priority than stickiness. In my experience, fluorescent proteins that behave as monomers in mammalian cells include: mTurquoise2, mEGFP, mNeonGreen, mScarlet(-I), and mCherry.
Keep the linker sequence short
In the early days of GFP applications, many were concerned with steric hindrance by the fluorescent protein. Thus, scientist began using relatively long linkers between the protein of interest and the fluorescent protein. These were either (i) coded by the multiple cloning site and therefore consist of random amino acid sequences or (ii) designed to form an inert, unstructured peptide and therefore consisted of glycines, serines and other small, non-aliphatic amino acids. However, long linkers increase the chance that a fusion protein is cleaved by proteases.
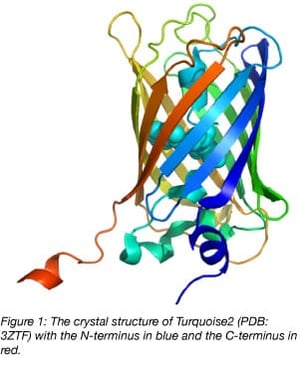
Nowadays, with ample structural information available, it’s clear that the C-terminus of fluorescent proteins derived from Aequorea victoria GFP sticks out and can be considered a linker (figure 1). In fact, for several FRET biosensors (yellow cameleon 3.60, EPAC, and Galphai), the C-terminus and N-terminus of the donor fluorescent protein can be truncated to increase the FRET efficiency.
Creating fusions at the N- and C- termini of your protein of interest
 The most straightforward fusion protein is made by connecting the N- or C-terminus of the fluorescent protein and protein of interest (POI). To indicate the sequence, people often use terminology like “C-terminal fusion,” but I find this very confusing because it can mean that the fluorescent protein is at the C-terminus of the POI or that the POI is at the C-terminus of the fluorescent protein. I prefer to describe the different parts of the fusion from N- to C-terminus. For instance, Lifeact-mTurquoise2 (Plasmid #36201) or mScarlet-tubulin (Plasmid #85045).
The most straightforward fusion protein is made by connecting the N- or C-terminus of the fluorescent protein and protein of interest (POI). To indicate the sequence, people often use terminology like “C-terminal fusion,” but I find this very confusing because it can mean that the fluorescent protein is at the C-terminus of the POI or that the POI is at the C-terminus of the fluorescent protein. I prefer to describe the different parts of the fusion from N- to C-terminus. For instance, Lifeact-mTurquoise2 (Plasmid #36201) or mScarlet-tubulin (Plasmid #85045).
We often use restriction enzymes in the multiple cloning site for generating fusions. For the POI-fluorescent protein fusions, our experience is that a linker of 4 amino acids (PVAT, when we use the AgeI site in the multiple cloning site) is sufficient. For the fluorescent protein-POI fusions, we generate straight fusions, without any linker.
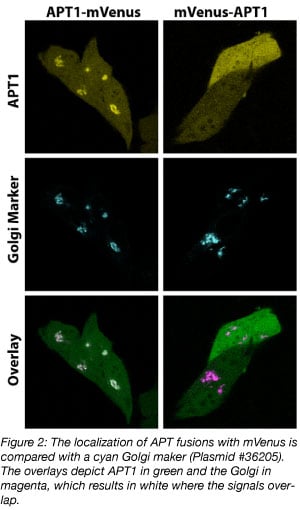
When both orientations are available, a direct comparison of these fusion proteins may reveal which of the two better preserves the function of the POI. For example, the APT1-mVenus fusion localizes at the Golgi, whereas the mVenus-APT1 fusion does not (figure 2). In the mVenus-APT1, a lipidation motif, which is needed for Golgi localization, is occluded and therefore this protein is mislocalized. The APT1-mVenus fusion is clearly a better choice to work with.
Inserting a fluorescent protein within your protein of interest
We have encountered several proteins for which the N- and C-terminus of the POI are necessary for its native properties. In these cases, an insertion of the fluorescent protein into the POI can work well since the N- and C-terminal residues of the fluorescent protein are relatively close together (figure 1).
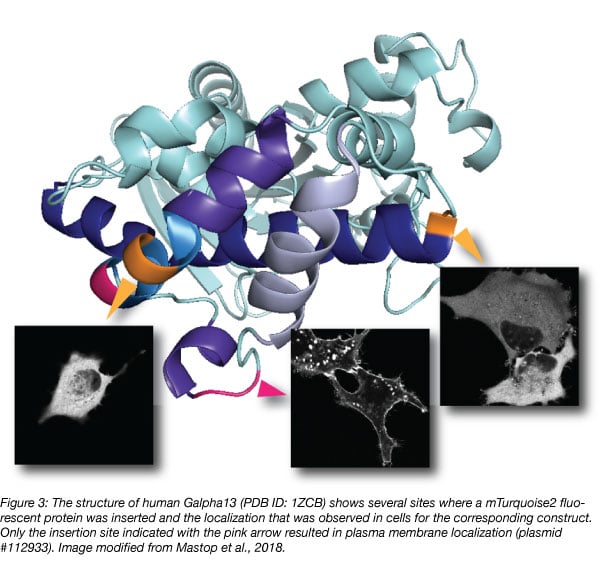
Where should the fluorescent protein be inserted? Sites that are less likely to disrupt the structure of the POI would work best. Ideally, structural information can guide this decision. In our experience, the best site for the insertion is a loop in between secondary structure (beta-sheets or alpha helices). An additional requirement is that the site of insertion does not interact with other proteins or biomolecules. Examples of the successful insertion of a fluorescent protein are the tagging of Ccd42 (Bendezú et al., 2015) and the alpha subunit of heterotrimeric G-proteins (Janetopoulos et al., 2001, Adjobo-Hermans et al., 2011).
Although structural information may guide design, it is advisable to generate multiple constructs with different insertions, since it is difficult to predict which one will work as illustrated in figure 3 (Mastop et al., 2018). Unbiased, random insertions can be generated with a transposon-based strategy (Sheridan, 2002).
Avoiding protein over-expression artifacts
The introduction of DNA that encodes a fusion protein adds protein to the existing pool of protein within a cell and may lead to over-expression artifacts. This is an important downside of the technology and should always be considered when interpreting results.
The recent revolution in genome editing methods (most notably CRISPR/Cas mediated HDR) enables the tagging of endogenous genes close to native expression levels. However, it is advisable to first express the fusion protein from a plasmid-based system to assess whether the chimera is still functional.
Final words on fluorescent protein fusions
There are several choices to consider before generating a fusion protein, e.g. linker length, the specific fluorescent protein variant, and where to add the fluorescent tag. These choices will determine how well the fusion reflects the function of the native protein that is tagged. The localization of the fusion protein can be verified and it can be compared to what is expected based on immunofluorescence or properties of the protein.
Determining other aspects of functionality (catalytic activity or interactions) can be challenging. If a fusion can complement a knock-down or knock-out of the corresponding protein, that is strong evidence that the fusion behaves well. But this is not conclusive evidence that the fusion operates in the same way as the native protein. Although a fluorescent protein fusion may never be able to act identical to the untagged protein, the benefits of being able to see your protein of interest usually outweigh potential negative consequences of the tag.
Many thanks to our guest blogger, Joachim Goedhart!
 Joachim Goedhart is an assistant professor at the Section of Molecular Cytology and Van Leeuwenhoek Centre for Advanced microscopy (University of Amsterdam). He develops, characterizes, and uses genetically encoded fluorescent probes. You can follow him on twitter: @joachimgoedhart.
Joachim Goedhart is an assistant professor at the Section of Molecular Cytology and Van Leeuwenhoek Centre for Advanced microscopy (University of Amsterdam). He develops, characterizes, and uses genetically encoded fluorescent probes. You can follow him on twitter: @joachimgoedhart.
References
Adjobo-Hermans, Merel JW, et al. "Real-time visualization of heterotrimeric G protein Gq activation in living cells." BMC biology 9.1 (2011): 32. PubMed PMID: 21619590. PubMed Central PMCID: PMC3129320.
Bendezú, Felipe O., et al. "Spontaneous Cdc42 polarization independent of GDI-mediated extraction and actin-based trafficking." PLoS biology 13.4 (2015): e1002097. PubMed PMID: 25837586. PubMed Central PMCID: PMC4383620.
Botman, Dennis, et al. "In vivo characterisation of fluorescent proteins in budding yeast." Scientific reports 9.1 (2019): 2234. PubMed PMID: 30783202. PubMed Central PMCID: PMC6381139.
Costantini, Lindsey M., et al. "Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions." Traffic 13.5 (2012): 643-649. PubMed PMID: 22289035. PubMed Central PMCID: PMC3324619.
Cranfill, Paula J., et al. "Quantitative assessment of fluorescent proteins." Nature methods 13.7 (2016): 557. PubMed PMID: 27240257. PubMed Central PMCID: PMC4927352.
Janetopoulos, Chris, Tian Jin, and Peter Devreotes. "Receptor-mediated activation of heterotrimeric G-proteins in living cells." Science 291.5512 (2001): 2408-2411. PubMed PMID: 11264536.
Klarenbeek, Jeffrey, et al. "Fourth-generation epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: characterization of dedicated sensors for FLIM, for ratiometry and with high affinity." PloS one 10.4 (2015): e0122513. PubMed PMID: 25875503. PubMed Central PMCID: PMC4397040.
Lambert, Talley J. "FPbase: A community-editable fluorescent protein database." Nature methods (2019): 1. PubMed PMID: 30886412.
Mastop, Marieke, et al. "A FRET-based biosensor for measuring Gα13 activation in single cells." PloS one 13.3 (2018): e0193705. PubMed PMID: 29505611. PubMed Central PMCID: PMC5837189.
Nagai, Takeharu, et al. "Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins." Proceedings of the National Academy of Sciences 101.29 (2004): 10554-10559. PubMed PMID: 15247428. PubMed Central PMCID: PMC490022.
Shaner, Nathan C., et al. "Improving the photostability of bright monomeric orange and red fluorescent proteins." Nature methods 5.6 (2008): 545. PubMed PMID: 18454154. PubMed Central PMCID: PMC2853173.
Sheridan, Douglas L., et al. "A new way to rapidly create functional, fluorescent fusion proteins: random insertion of GFP with an in vitro transposition reaction." BMC neuroscience 3.1 (2002): 7. PubMed PMID: 12086589. PubMed Central PMCID: PMC117241.
van Unen, Jakobus, et al. "A New generation of FRET sensors for robust measurement of Gαi1, Gαi2 and Gαi3 activation kinetics in single cells." PloS one 11.1 (2016): e0146789. PubMed PMID: 26799488. PubMed Central PMCID: PMC4723041.
Additional resources on the Addgene blog
- Read our fluorescent proteins blog posts
- Download the fluorescent proteins 101 eBook
- Read our plasmids 101 blog series
Resources on Addgene.org
- Browse the fluorescent protein collection
- Find empty backbones for fluorescent protein fusions
- Browse the fluorescent proteins plasmid kits






Leave a Comment