 You’ve prepped your DNA and you’re ready to get started on the next step of your experiment. But in many cases, you won’t see any signs of DNA in your final tube after purification. How do you know if you actually have DNA in your tube without seeing it?
You’ve prepped your DNA and you’re ready to get started on the next step of your experiment. But in many cases, you won’t see any signs of DNA in your final tube after purification. How do you know if you actually have DNA in your tube without seeing it?
There are many ways to do this and the method you choose could be based on your downstream application, time, and instrument availability.
UV absorbance
Using UV absorbance is one of the most common ways to quantify DNA. This method involves measuring the absorbance/transmission of light through a liquid to determine the concentration of substances in the liquid. Molecules absorb different wavelengths of light to different degrees and many molecules have a specific wavelength that they maximally absorb. The spectrophotometer measures these absorbances using UV-transparent cuvettes. First you measure the absorbance of the buffer that the DNA is in. This is a “blank” and measures the background absorbance. Then you measure the absorbance of the DNA sample.
These absorbance measures give you an idea of the concentration of your DNA prep and whether there are any other contaminants. Nucleic acids (DNA and RNA) absorb maximally at 260 nm. Proteins on the other hand absorb best at 280 nm and organic compounds and chaotropic salts maximally absorb at 230 nm. The A260/A280 ratio is used as an indicator of DNA purity. Ideally, this number should be between 1.8 and 2.0. The A260/A230 ratio is best if greater than 1.5.
Then, using the A260 reading, you can calculate the DNA concentration. Generally, A260 of 1.0 is equivalent to 50 ug/ml pure dsDNA. Use the following formula to estimate your DNA:
Concentration (ug/ml) = A260 reading x dilution factor x 50 ug/ml
This method is quick and simple and doesn’t require any special reagents. However, there’s limited sensitivity at low concentrations of DNA and it can’t distinguish between DNA and RNA.
See our protocol about using UV absorbance to quantify DNA
Fluorescence dyes
Another way to quantify DNA would be to use fluorescent dyes that fluoresce when bound to DNA. The main distinction here is that these dyes, such as PicoGreen or SYBRGreen, are specific for double strand DNA (as compared to spectrophotometric methods that measure all nucleic acids). These methods are more sensitive than UV absorbance, especially when you expect low concentrations in your samples and are often used to quantify DNA for next generation sequencing.
Unlike absorbance-based methods, fluorescence-based methods require a standard curve, a set of samples with a known DNA quantity and their corresponding fluorescence. That way, you can compare the fluorescence of your sample against this curve to quantify your DNA prep. Though this method takes more time to set up in the lab, you might not have to do the calculation yourself as many fluorometers will calculate your sample concentration for you.
Agarose gel electrophoresis
Though not the quickest way to quantify DNA, you can use the agarose gel method to not only find out how much DNA you have, but also to see whether your DNA is intact or the correct size. This is something that absorbance based methods can’t tell you and you don’t need a spectrophotometer or a fluorometer to quantify DNA this way.
First, start by pouring your gel containing a DNA intercalating dye (ex: ethidium bromide) and choosing a DNA ladder with known concentrations. Many commercial DNA ladders will tell you the concentration of each band in the ladder. You then run samples of your DNA at different dilutions and quantify based on band intensities of your DNA relative to the intensity in the ladder. It’s best to compare the band intensity of the fragment in the ladder that is closest in size to your piece of DNA. This method is best used for DNA fragments (like a PCR product). This method also gives an indicator of DNA or RNA contamination based on the presence of other bands or streaking. Unlike absorbance based methods, this method doesn’t tell you about contaminating proteins or chaotropic salts.
Capillary electrophoresis
Quantifying DNA using capillary electrophoresis is similar to quantifying DNA using gel electrophoresis. Except, it’s smaller. And automated.
Unlike gel electrophoresis, you only need 1-2 ul of sample and the run time is just a few minutes per sample. During the run, DNA fragments move through a micro- or nanofluidic channel and the sample is separated by electrophoresis. Smaller fragments migrate quicker than larger fragments. These fragments are quantified over time using a fluorescent dye that intercalates into the DNA such that smaller fragments are quantified first before larger fragments.
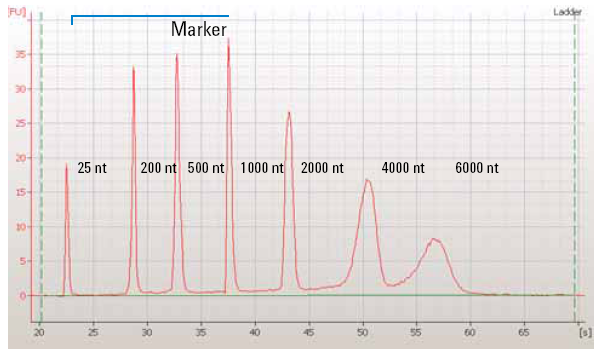 |
| Figure 1: The quantification of a RNA ladder using capillary electrophoresis which shows fluorescence units over time. Smaller fragments migrate more quickly than larger fragments. Image from OpenWetWare. |
This method is often used preceding next-generation sequencing or microarray studies and is not used as much for the standard plasmid prep.
Diphenylamine method
Another absorbance based method for quantifying DNA uses diphenylamine. Diphenylamine reacts with deoxyribose sugars under acidic conditions and forms a blue complex that can be quantified at 595 nm.
This method is time consuming and has low sensitivity so it’s not used much. However, the measurements are made in the visible range and can be read with a standard ELISA reader if no other instruments are available.
Some last reminders about quantifying DNA
In choosing your DNA quantification method there’s many things to consider: cost, time, equipment, and expected DNA concentration. It’s important to consider the advantages and disadvantages of each method and when one method might be more suited than another.

Additional resources on the Addgene blog
- Read more about molecular biology protocols and tips
- Find the best way to elute and store your plasmid DNA
- Learn about the different grades of plasmid DNA preps
Resources on Addgene.org





Leave a Comment