This post was contributed by Jake Watson and Javier García-Nafría from the MRC Laboratory of Molecular Biology.
Plasmid cloning is an essential part of any molecular biology project, yet very often, it is also a bottleneck in the experimental process. The majority of current cloning techniques involve the assembly of a circular plasmid in vitro, before transforming it into E. coli for propagation. However, while not widely known, plasmid assembly can be achieved in vivo using a bacterial recombination pathway that is present even in common lab cloning strains.
This intrinsic bacterial recombination pathway, referred to as recA-independent recombination, joins together pieces of linear DNA through short homologous sequences at their termini, and likely functions as a bacterial DNA repair mechanism. The pathway is ubiquitous, with successful recombination reported in all laboratory E. coli strains tested so far.
The use of this pathway as a cloning tool was first reported over 25 years ago (Douglas and Howard, 1991, Bubeck et al., 1993). But why did this technique fail to gain traction? It is likely that both vector amplification with low fidelity polymerases and the cost of synthesizing longer oligos, which could be avoided using restriction enzyme cloning, were more prohibitive at the time. With these limitations no longer an issue, bacterial in vivo cloning is now able to come of age.
The in vivo assembly cloning protocol
In Vivo Assembly (IVA) cloning (Garcia-Nafria et al., 2016) uses the bacterial recombination pathway to allow any cloning procedure to be performed using a simple two-step, 2 hr protocol prior to transformation (Figure 1). As this approach requires no special reagents or purification kits, it is low cost and can be immediately adopted by any laboratory.
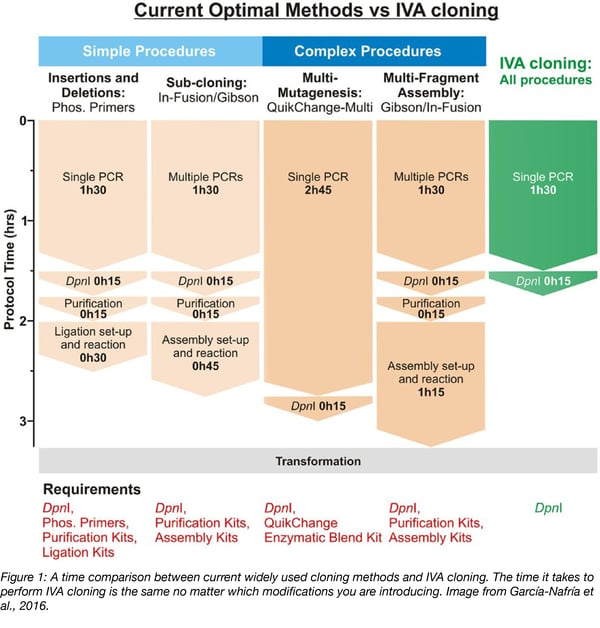
All procedures are performed using the same three step protocol:
- Fragment generation - Any linear DNA fragments can be assembled in vivo as long as they have homologous sequences at their termini. It doesn’t matter if the DNA is PCR amplified, restriction digested or even a synthesized gene. PCR is the primary method to produce the linear fragments, as desired modifications and homologous regions can be encoded in the primer sequences. Multiple plasmids can be included in a single PCR, for example for subcloning genes from different templates. All PCRs are run as an 18 cycle, 25 μl single-tube reaction. If you are synthesizing a new DNA fragment, encode homology arms to allow immediate co-transformation with linearized vector backbone.
- DpnI digestion - 1 μl DpnI is added to the PCR mix to remove parental DNA (37 °C for 15 mins). This enzyme specifically cuts methylated DNA, so it will selectively destroy the template, but not newly synthesized sequences, limiting the number of false positive colonies.
- Transformation - the linear fragments are transformed into E. coli and plasmid assembly (re-circularization) occurs in vivo. We use XL-10 Gold E. coli, but have successfully tested DH5a, TOP10, and Mach1. No common lab cloning strain has failed so far. What is most important is the transformation efficiency of the strain: when asking the cell to perform multiple recombination events, all linear fragments need to get into the same cell. For simple cloning requiring just one or two recombination events, homemade competent cells of 107 CFU/μg are perfectly fine, yet performing multiple modifications will require ultracompetent (109 CFU/μg) cells.
Primer design
Using specific primer design (Figure 2), IVA cloning can be used to perform any plasmid modification, from insertions, deletions, and point mutagenesis to complex assembly of multiple genes. Primer design for each type of modification is shown in Figure 2, and is discussed in more detail below. The main requirement for IVA cloning is carefully designed primers. The majority of mistakes are due to incorrect primer sequences!
Primers consist of two regions: the 3’ end binds template DNA for PCR amplification, while modifications and homologous sequences are encoded at the 5’ end. First design the template binding region, with a melting temperature (Tm) of around 60 °C for all primers. The length of the homologous region should be around 20 bp, but pay attention to the sequence’s Tm, as a higher Tm give more efficient recombination. We normally use a Tm between 47-55 °C for the homologous region (calculated using Oligo Calculator).
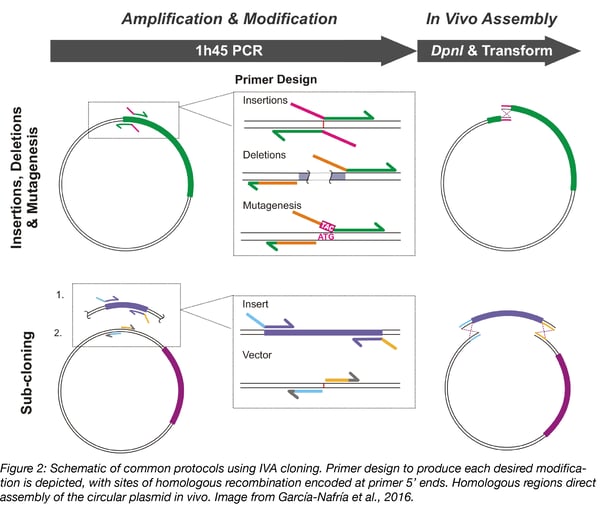
Insertions - To insert short sequences of up to 100 bp, such as epitope tags, design primers with template binding regions to amplify away from the insertion site. Encode half the new sequence at the 5’ end of each primer, with an overlap for homologous recombination.
Deletions - To remove part of a plasmid, order primers that bind at either side of the undesired portion, designed to amplify the rest of the vector sequence. Encode homology in the forward primer by adding a short sequence complementary to the 5’ end of the reverse primer.
Mutagenesis - Mutagenesis is performed by designing primers astride the old codon, amplifying the whole vector. The new codon and homologous region can be included at the 5’ end of either primer.
Subcloning - To move a gene from one vector to another simply amplify the vector and insert with separate primer pairs in the same PCR. Add homologous sequences to one of the primer pairs to drive assembly.
Combining multiple modifications
A key feature of IVA cloning is the ability to combine multiple modifications. Any IVA primers can be used in combination to simultaneously perform modifications, simply by adding all the necessary primers to the same PCR mix. Pairs of primers will amplify the whole vector sequence between modification sites (see Figure 3), producing multiple fragments with specific homologous ends. These fragments are assembled in vivo to produce the multiply modified product. For example, by combining multiple primers, a C-terminal epitope tag can be moved to the N-terminus of a protein-coding sequence, or a mutation can be introduced in a gene, while adding a GFP-tag (Figure 3).
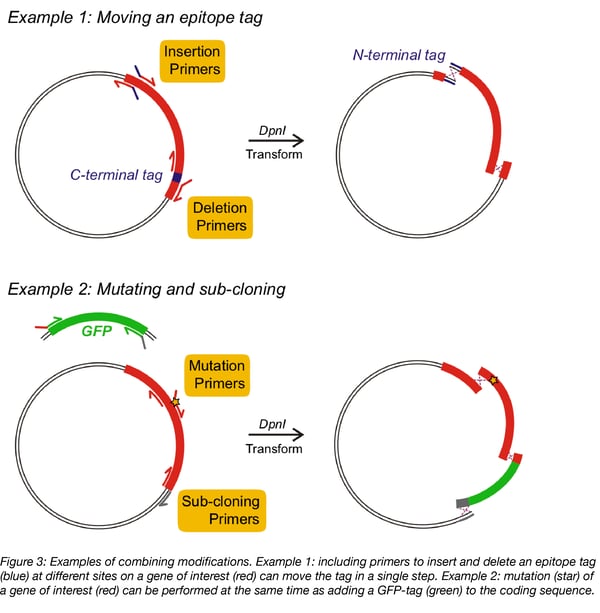
Achieving multiple modifications requires multiple recombination events to assemble the final plasmid. At least 5 fragments can be correctly assembled using IVA cloning, although the efficiency of assembly decreases as the cloning complexity increases. The efficiency of recombination is increased with greater homology, so performing more complex procedures can be aided by extending homologous sequences (to 25-30 bp).
Tips and tricks for IVA cloning
Use a sequence manager
A sequence manager, such as SnapGene, can be very helpful to confirm primer design.
Use minimal template DNA
Use 1 ng of PCR template DNA to minimize false positive colonies. We routinely screen just two colonies for simple cloning procedures.
Use high fidelity DNA polymerase
This will minimize random errors during PCR: we recommend Phusion or Q5.
Speed up your life
Rather than making up your PCR mix every time, pre-made aliquots of PCR master-mix, containing buffer, polymerase and dNTPs, can be stored frozen: just defrost and add your primers and template.
Amplifying the ‘unamplifiable’
Check your amplification by gel electrophoresis. In our experience, good amplification after PCR means successful plasmid assembly (if primer design is correct). Some DNA sequences are not easy to amplify by PCR, so we usually add DMSO (3%) and Betaine (1 M) to the PCR mix.
When PCR is not possible
Some plasmids just won’t amplify! Examples of this are the GC-rich ITRs of AAV genomic DNA encoding plasmids, or promoters such as the chicken beta-actin promoter of the pHLsec vector. In this scenario, IVA can be employed by cutting the vector with restriction enzymes and adding the corresponding homologous sequences to the insert by PCR.
Many thanks to our guest bloggers Jake Watson and Javier García-Nafría from the MRC Laboratory of Molecular Biology.
 Jake Watson is a postdoctoral researcher in neurophysiology at the MRC Laboratory of Molecular Biology. He is currently investigating the molecular biology of synaptic transmission and plasticity.
Jake Watson is a postdoctoral researcher in neurophysiology at the MRC Laboratory of Molecular Biology. He is currently investigating the molecular biology of synaptic transmission and plasticity.
 Javier García-Nafría is an investigator scientist at the MRC Laboratory of Molecular Biology. He is interested in neuronal signal transduction, and uses electron cryo-microscopy to understand receptor function.
Javier García-Nafría is an investigator scientist at the MRC Laboratory of Molecular Biology. He is interested in neuronal signal transduction, and uses electron cryo-microscopy to understand receptor function.
Follow them on Twitter @jakefwatson and @JGarciaNafria.
References
1. Jones, Douglas H., and Bruce H. Howard. "A rapid method for recombination and site-specific mutagenesis by placing homologous ends on DNA using polymerase chain reaction." Biotechniques 10.1 (1991): 62-66. PubMed PMID: 2003926.
2. Bubeck, Peter, Monica Winkler, and Wilfried Bautsch. "Rapid cloning by homologous recombination in vivo." Nucleic acids research 21.15 (1993): 3601. PubMed PMID: 8346047. PubMed Central PMCID: PMC331480.
3. García-Nafría, Javier, Jake F. Watson, and Ingo H. Greger. "IVA cloning: a single-tube universal cloning system exploiting bacterial in vivo assembly." Scientific reports 6 (2016): 27459. PubMed PMID: 27264908. PubMed Central PMCID: PMC4893743.
Additional resources on the Addgene blog
- Read all of our Plasmids 101 blog posts
- Read our blog post on 5 factors to consider before choosing a cloning method
- Check out all of our blog posts on plasmid cloning
Resources on Addgene.org
- Read our protocols for plasmid cloning
- Find our molecular biology reference here
- Find molecular biology protocols here

Topics: Plasmid Cloning, Molecular Biology Protocols and Tips, Plasmids





Leave a Comment