Originally published Aug 3, 2017 and last updated Apr 6, 2021 by Will Arnold.
Introducing NGS in 2017 for all incoming plasmids was a big change for Addgene - we had used Sanger sequencing for quality control for over ten years. Now, thanks to a generous donation by the Hirsh family, we are able to take greater ownership of this process with in-house sequencing! In addition to sequencing plasmids, we have also started using NGS to check the quality of other services that we provide, including viral preps and pooled libraries.
As anyone who has ever worked to sequence DNA on NGS platforms will tell you, while the process has become much more refined and user friendly, to own the process entirely remains a large undertaking. Priority number one was to ensure that this changeover had no impact on our ability to provide requesting scientists high quality data!
Addgene's new in-house NGS process
To start, the process is only made possible by the use of our high-throughput DNA isolation process that yields high-quality isolated DNA samples of sufficient quantity for sequencing. This process is completed in a plate format yielding anywhere from two to six plates of 96 samples per week. This is where our new process will begin. Partnering with seqWell, we are using the plexWell technology to easily and quickly create Illumina sequencing libraries. From start to finish the library preparation process only takes about one day, even for six plates (576 individual plasmids)! These libraries are then QC’d, pooled, and prepared for Illumina sequencing on our newly donated MiSeq.
For Complete Plasmid Sequencing we perform a 2x251 run on our MiSeq that takes about two days to complete. After the run completes, we begin our assembly process. Again, thanks to our partners at seqWell we make use of a pipeline that takes the raw data from each sample in our pool and individually assembles the reads into a single FASTA sequence that our QC scientists can easily analyze (see below for what we look out for while analyzing the plasmid sequences).
As with all things at Addgene, each part of this new process involves a significant amount of QC. First, we perform Picogreen quantification on each and every isolated plasmid sample and normalize each to fall within a 10 fold range. Plasmids that don't meet the lower bound of this range are “failed” and removed from the plate to be prepped again. These holes are then filled with other samples in our queue such as viral samples for Viral Genome Sequencing (VGS). QC is performed throughout the library preparation process and includes Picogreen quantification of various intermediate steps, a sizing electrophoretic gel, and qPCR quantification of the final libraries.
A broader view of the whole plasmid
To confirm the sequence of the plasmid, we examine three things:
- Aligning the NGS result to a reference sequence to confirm backbone elements.
- Confirming the gene/insert by aligning to NCBI entry or using BLAST.
- Confirming tags and fusion proteins.
We’ll break down each of these steps below.
Aligning the sequence
The first step we take when performing quality control of newly deposited plasmids is to align the NGS result to a reference sequence like a known backbone sequence. We’re not necessarily expecting a perfect match - we will often find a few mismatches in the origin of replication or other common backbone elements. Since we’ve successfully grown the plasmid in culture to prepare it for sequencing, we feel confident that these few minor mismatches usually don’t affect the function of the plasmid.

Confirming the insert
We usually confirm the insert through BLAST or by direct alignment to an NCBI reference sequence or a sequence provided by the depositor. Depositing labs often provide insert sequences or annotated Genbank files that are useful for more complicated plasmids, like those that contain synthesized regions that have no publicly available reference sequence or plasmids containing genes with many modifications. We look for point mutations, truncations, and insertions that could compromise function. When we do find mutations, we check to see if they affect the translated amino acid. We also confirm that the species of the gene matches the data associated with the plasmid.
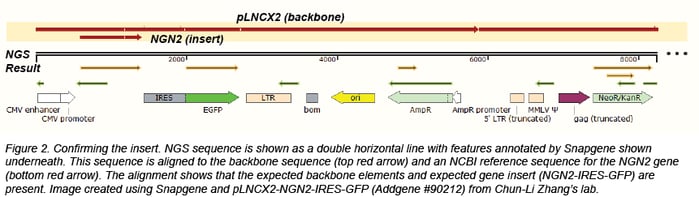
Confirming tags and fusion proteins
Finally, we confirm promoters, tags, fusion proteins, and selectable markers by detecting common features using Snapgene. If we find information that differs from what we would expect given the data provided by the depositing lab, we call these “quality control (QC) issues.” We then ask the depositing laboratory to review the discrepancies. If the depositing laboratory confirms that these differences are expected and do not affect plasmid function, we will update information on the plasmid’s page.
Sometimes we’re missing a piece of the plasmid
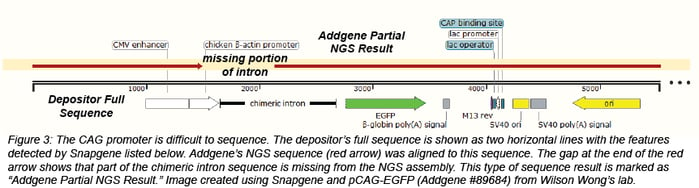
There are regions of some plasmids that are particularly difficult to sequence and assemble, including GC rich regions. For example, the CAG promoter, a hybrid promoter consisting of a CMV enhancer, chicken beta actin promoter, and rabbit beta-globin splice acceptor, contains sequence that is over 80% GC. Some IRES sequences also contain regions that are GC rich. Since NGS assembles a larger sequence from many smaller sequences, regions with repeats may not assemble correctly. Due to these issues, NGS for some plasmids will not result in one complete, circular assembly. Most of the time, these plasmids are returned as one or two partial assemblies. If we obtain an NGS result that is useful, but not 100% complete, we will still make this data available as a partial sequence with the heading "Addgene Partial NGS Result." In some cases, there will be more information about our sequencing results in the Depositor Comments section at the bottom of the plasmid page.
Viral preps and pooled libraries
In addition to regular plasmid sequencing, many of you will be familiar with our efforts to ensure access to high quality, ready-to-use viral preps and pooled libraries.
Viral samples are filled into the same process as our normal Complete Plasmid Sequencing (CPS) workflow and yield sufficient data to capture the entire viral genome. The full details of that workflow and its analysis can be found here: A Novel Next-Generation Sequencing and Analysis Platform to Assess the Identity of Recombinant Adeno-Associated Viral Preparations from Viral DNA Extracts.
Pooled libraries are a bit more challenging to QC by NGS as they are often quite different from one library to another. They also differ from the CPS process in that we do not fully sequence all plasmids in the pool but instead selectively sequence the variable portion of the plasmid in the pool - ex. the gRNA in a CRISPR library. In general, our QC scientists work with the depositing lab to develop a PCR strategy suitable for the library in question. We will order primers that can amplify the insert of the library. This PCR step also adds the necessary adapter sequences for Illumina sequencing as well as unique barcodes per sample so we can assign sequencing reads to the correct sample. The PCR is then performed, optimized as needed, and cleaned up using a standard PCR cleanup kit to remove any template DNA and primers.
After performing extensive QC on that sample, we’ll then load it on to our new MiSeq and set up the run making sure to note the appropriate barcode for each sample. About 24 hours later we unload the FASTQ data from our instrument and begin the analysis. We use a modified version of the Python program described by Joung et al. 2017, which we hope to make available as part of our OpenBio Repository sometime soon. Stay posted!

References
Guerin K, Rego M, Bourges D, Ersing I, Haery L, Harten DeMaio K, Sanders E, Tasissa M, Kostman M, Tillgren M, Makana Hanley L, Mueller I, Mitsopoulos A, Fan M (2020) A Novel Next-Generation Sequencing and Analysis Platform to Assess the Identity of Recombinant Adeno-Associated Viral Preparations from Viral DNA Extracts. Human Gene Therapy 31:664–678 . https://doi.org/10.1089/hum.2019.277
Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F (2017) Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12:828–863 . https://doi.org/10.1038/nprot.2017.016
Topics: Plasmids 101, Addgene News, Plasmids





Leave a Comment