 This post was contributed by guest blogger Joe Mellor, Founder and CEO of seqWell Inc.
This post was contributed by guest blogger Joe Mellor, Founder and CEO of seqWell Inc.
Plasmids and PCR products are the bread and butter of molecular biology labs the world over. Scientists have traditionally used Sanger sequencing to validate these constructs, as the relatively low cost and quick turn-around time of Sanger sequencing have historically matched the needs of most molecular biology labs. Recent and rapid advances in technologies that permit large-scale creation and synthesis (“writing”) of longer pieces of synthetic DNA, as well as the advent of extremely fast, cheap and accurate sequencing (“reading”) of DNA, have changed our collective thinking about the feasible size and scope of projects in many labs. However, the high costs of sample preparation for high-throughput next generation (NGS) sequencing have prevented laboratories from using these methods for routine processes like plasmid validation.
At seqWell, Inc., our mission is to overcome crucial challenges in NGS by developing technologies that can help unlock the potential of modern sequencing instruments by enhancing the efficiency and simplicity of library prep. As part of our mission, we’ve been working with Addgene to develop and apply our plexWell™ Library Preparation Technology for NGS-based sequencing and confirmation of Addgene’s large and growing collection of curated plasmids from all over the world. The rest of this piece will describe plexWell™ in more detail, and how we are using this technique in our partnership with Addgene to sequence large numbers of plasmids.
plexWell™ technology overview
For any NGS experiment, the DNA sample being sequenced must be sheared into smaller fragments that are then barcoded for identification. This mixture of barcoded fragments is known as a *library*. Parallel sequencing of the fragments within a library allows researchers to reconstruct the full individual sequence. Traditional NGS workflows are limited by costly library prep, as each sample requires an individual library.
The plexWell™ workflow is a sequential “pooled library prep”, in which a relatively large number of samples are subjected to a DNA-barcoding step that uniquely labels DNA from each well of a 96-well plate. Then, samples from several plates are pooled together to create a single library containing fragments carrying sample-specific barcodes. Rather than continuing to process 100s to 1000s of samples individually, plexWell™ reduces every 96 well plate to a single tube after the first tagging step. As an additional benefit of plexWell™’s sequential tagging procedure, the quantities of library molecules for each sample remain highly normalized across a wide range of input DNA concentrations. This process is explained more fully in the animated Fig. 1 below.
Normalization is a fundamental and often understated problem in multiplex NGS library preparation. The challenge here is that, as more samples are sequenced simultaneously, it gets harder and harder to obtain balanced and sufficient coverage of all samples. The root of the normalization problem often arises from the fact that large numbers of individual input DNA samples (e.g. a collection of 96 plasmids on a plate) will typically have a wide range of input quantities. This variation is problematic for conventional NGS library prep methods because the yield or quality of individual libraries can be influenced by the quantity or quality of input samples.
To minimize these issues, researchers usually include a separate normalization step where individual input DNA samples are brought in line with each other before proceeding to library prep. Even with this “pre-prep” normalization, it is also often the case that individual libraries prepared from multiple samples will then also need to be quantified and re-normalized in order to be combined with other samples in a single multiplexed pool. The cost of these normalization steps, measured in time and consumables, can be daunting for large numbers of samples.
As mentioned above, one of the primary technical benefits of plexWell™ is that normalized libraries are produced from large numbers of samples with a wide range of input concentrations (via proprietary chemistry). This turns out to be critically important for getting adequate sequencing depth and coverage in order to, for example, assemble hundreds of different plasmids from a single Illumina MiSeq® run. As shown in Fig. 2, below, differences in input concentration across a 100-fold range (1ng – 100ng) are normalized to two-fold variation above and below mean read count per sample, and the amount of information per read does not vary by DNA input amount.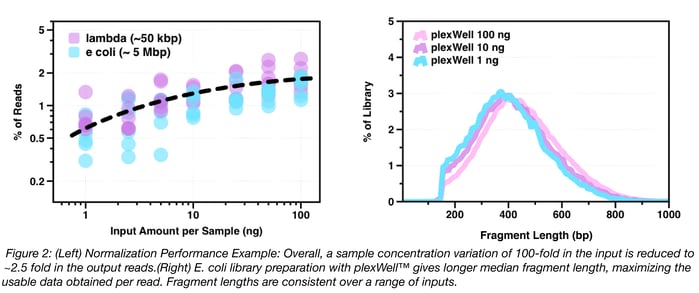
plexWell™ + Addgene = high quality NGS-assembled plasmids
Because of the enhanced normalization of read count and fragment length across samples, NGS data obtained via plexWell™ libraries is well suited to large-scale de novo assemblies of short constructs like plasmids. The plexWell™ workflow also offers a highly efficient NGS-based alternative to Sanger sequencing because the sample identity information is always displayed at the individual read level, and because custom primers or "primer-walking" reactions are completely unnecessary for sequencing multi-kb DNA inserts and larger targets. The NGS readout also reveals additional information about the presence of non-plasmid DNA or heterogeneous and/or mixed clones present in the original sample.
At seqWell, we have worked with Addgene to develop a highly optimized bioinformatics pipeline that will return high-quality assembled and circularized plasmids from plexWell™ NGS data. Once Addgene produces plates of plasmid DNA, they are shipped to seqWell, where they are prepared as plexWell™ libraries, sequenced on an Illumina® sequencer, and then analyzed with our plasmid assembly pipeline. To date, this has resulted in high quality NGS-based assemblies for over 3000 plasmids in the Addgene collection.

Many thanks to Joe Mellor, our guest writer from seqWell Inc.
 Joe Mellor is the Founder and CEO of seqWell, Inc. He is a sequencing technologist and computational biologist with a passion for solving critical sequencing problems. He received his PhD in Bioinformatics from Boston University, and did postdoctoral work at Harvard Medical School and the University of Toronto. He is an author of 20+ scientific publications and patents.
Joe Mellor is the Founder and CEO of seqWell, Inc. He is a sequencing technologist and computational biologist with a passion for solving critical sequencing problems. He received his PhD in Bioinformatics from Boston University, and did postdoctoral work at Harvard Medical School and the University of Toronto. He is an author of 20+ scientific publications and patents.

Topics: Addgene News





Leave a Comment