Immunoprecipitation (IP) uses immobilized antibodies, or immunoglobulins, to isolate a specific protein out of a complex mix. Using this technique, users can look for the presence or absence of a protein, determine if a protein is up or downregulated, examine a protein’s stability or post-translational modifications, or study how a target protein interacts with other proteins or nucleic acids. Read on to learn more about this versatile technique.
Immunoprecipitation Overview
An immunoprecipitation reaction is typically carried out in one of two ways. Both methods utilize the same basic strategy: immobilize the antibody or antibody-target protein immune complexes, wash away unbound protein, elute and measure the target. However, the two different approaches allow for optimized strategies for differing amounts of target proteins or antibody bonding strengths.
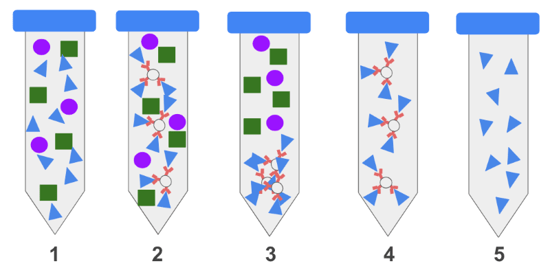 . . |
|
Steps of an IP: 1) Protein extracts from cells or tissues contain a complex mix of proteins seen as green squares, purple circles and blue triangles. 2) An antibody immobilized on a bead binds specifically to the blue triangle protein but not the other. 3) The beads are collected by centrifugation or a magnet. 4) Unbound proteins are washed away. 5) The target protein is eluted. |
In the first method, an antibody against a target protein is immobilized, or tethered, on agarose or magnetic beads and then incubated with a protein mix. During the incubation the immobilized antibody binds to the target protein, thereby tethering it to the beads. Unbound proteins in the mix are removed through a series of wash steps and the target protein is then eluted. This method is appropriate for most situations.
A slightly different method is desirable if the antibody is expected to bind weakly to the target protein or if the target is present in low amounts. In this method, the free antibody is incubated with the protein mix and antibody-target protein immune complexes are allowed to form. Following incubation, beads are used to immobilize the antibody-protein complexes, and as in the first method, unbound proteins are washed away and the target protein eluted.
Immobilization of the primary antibody
Immobilization is the key to immunoprecipitation and simply refers to the process of anchoring an antibody, often called the capture antibody, to agarose or magnetic beads in a way that also allows the antibody to bind to the target protein. One of the most common immobilization methods utilizes bacterial cell wall proteins, Protein A and Protein G, which bind to the fragment crystallizable region (Fc) of antibodies with a high affinity (Hjelm, 1972, Bjorck 1984). Protein A, Protein G, or the recombinant Protein A/G are conjugated to agarose or magnetic beads and allowed to bind to the antibody being used for IP. These proteins bind only to the Fc portion of the immunoglobulin, leaving the antigen binding sites free to capture the target protein. The beads can be collected by centrifugation (agarose beads) or a magnet (magnetic beads), providing a solid support while unbound proteins are washed away.
While both Protein A and Protein G have an affinity for immunoglobulins, the strength of the interaction varies between antibody host species and isotypes. Consequently, users need to choose the protein type that is most compatible with the host species and isotype of their primary antibody. To circumvent this, a recombinant protein, Protein A/G, was developed, which can be used for most antibodies compatible with either Protein A or G.
While commonly used, Protein A and Protein G are not suitable for all applications, such as protein isolation from serum. In this case, Protein A and Protein G will bind both the primary antibody and serum immunoglobulins indiscriminately, causing competition for binding sites. In cases like these, antibodies can be directly conjugated to agarose or magnetic beads with commercially available chemical agents. Directly conjugating the primary antibody has the additional benefit of permanent linkage. This means that the primary antibody will not co-elute with the target protein, eliminating the risk of the primary antibody interfering with downstream assays. This is particularly advantageous if the antibody used for IP is derived from the same species as the antibody that will be used for detection in a subsequent Western blot.
When choosing beads, there are several factors to consider. Agarose beads are sponge-like and vary in shape and structure. Their porous surface provides a large area for binding, but antibodies conjugated within the pores are often inaccessible to their targets. Magnetic beads are solid spheres and tend to be much smaller than agarose beads. Proteins conjugate on the surface of magnetic beads, and are therefore completely accessible. The yields from magnetic beads are equivalent, if not slightly higher, than those of agarose beads, despite the size difference. Magnetic beads are particularly advantageous because they do not require centrifugation between washes which can cause loss of beads, break weak protein interactions, and limit high throughput procedures. However, agarose beads are often a more cost-effective option.
Target protein/antibody interactions
To prepare for an IP, samples are typically lysed in a non-denaturing buffer containing non-ionic detergents, such as NP-40 or triton-X, to preserve native protein conformations and interactions. For difficult to release proteins, including those in the nucleus, more stringent buffers may be needed. Since many factors, including salt concentration, ionic strength and pH, affect binding the lysis buffer may need to be adjusted depending on the sample. It is important to remember that sample lysates will also contain proteases and phosphatases that may degrade the target protein. To prevent this, include protease and phosphatase inhibitors in the lysis buffer, keep samples on ice, and perform the IP at 4oC.
When choosing the capture antibody for IP, be sure to choose one that recognizes the target protein in its native conformation. Frequently, the antibodies used for a standard, denaturing Western blot will not be suitable for an IP, as proteins in an IP are not denatured (learn more about antibodies here!)
When using Protein A/G to immobilize the antibody, it is recommended to preclear the sample to remove any proteins that may interact nonspecifically with the capture antibody or Protein A/G. To preclear, incubate the sample as you would in an IP using a nonspecific antibody from the same host species as the capture antibody. This will deplete the nonspecific proteins from the sample prior to beginning the IP.
During the IP, the target protein in the sample binds to the capture antibody and is immobilized. The ideal antibody concentration to use will vary between capture antibodies and can be determined through titration. Unbound or weakly bound proteins in the sample are removed during a series of wash steps following the incubation step. The wash buffer has an optimal pH and ionic strength that breaks weak interactions from residual non-specific proteins but leaves the target protein/capture antibody intact. The wash step is repeated several times to ensure non-specific proteins are completely removed. Wash steps should be performed even with a pre-cleared sample.
Eluting the target protein for downstream testing
Once the non-specific proteins are removed through washing, the target protein can be eluted. The elution method will vary depending on the specific target protein:capture antibody interaction and/or the downstream application. Typically, if the target protein will be detected in a Western blot then it can be eluted by boiling the beads in SDS. (To learn more about Western blotting check out our blog post The Basics of Western Immunoblotting.) For mass spectrometry, elute in a urea-containing lysis buffer. Alternatively, the protein can also be eluted using a low pH buffer such as 0.1M glycine pH 2.5 and immediately neutralized with Tris pH 8-8.5. Low pH glycine is very effective at disrupting the antibody antigen complexes without permanently altering the structure of the protein.
Controls
As with any experiment, it is critical to include proper positive and negative controls for an IP. When possible, perform the IP in parallel with both a sample known to express the protein of interest, such as a sample transiently transfected or stably infected with the protein of interest and a sample that does not. Common negative controls include knockout cell lines or tissues that do not express the protein. With this control set, you should see your target protein IP’d in the positive control sample but not in the negative control, confirming that the capture antibody is functioning as expected. In order to identify any non-specific interactions, perform parallel IPs with beads only and an isotype control antibody.
Alternative applications
Oftentimes, an IP-appropriate antibody simply isn’t available for a specific target. In cases such as this, an epitope such as c-Myc, GFP or V5 can be used to tag the protein of interest on the C- or N-terminus. In this variation, called a pull-down assay, an antibody against the tag is used to isolate the protein of interest. The wide availability and high specificity of anti-epitope antibodies make this approach appealing, but the disadvantages must be carefully considered before using. For example, the tag may affect the confirmation of the target protein or interfere with protein interactions of interest. Tagged proteins are typically expressed at much higher levels than endogenous proteins. Data obtained from experiments using a tagged protein may not be translatable to the endogenous system and will need to be verified using alternative methods.
Finally, IP can also be used to study proteins that interact with nucleic acids. Labs studying epigenetics routinely use the chromatin IP (ChIP) technique to identify DNA binding proteins involved in histone modification. Similarly, RNA IP (RIP) can be used to isolate proteins that bind to RNA, though the details of these methods are beyond the scope of this post.
We hope that this blog has provided you with a useful overview of the IP method. If you found this blog helpful be sure to check out Addgene’s blog for other antibody topics.

References and resources
References:
Björck L, Kronvall G (1984) Purification and some properties of streptococcal protein G, a novel IgG-binding reagent. J Immunol 133(2):969-974.
Hjelm H, Hjelm K, Sjöquist J (1972) Protein a from Staphylococcus aureus. Its isolation by affinity chromatography and its use as an immunosorbent for isolation of immunoglobulins. FEBS Letters 28:73–76
Additional resources on the Addgene blog:
- Introduction to Immunofluorescence
- View all of Addgene's antibody posts
Resources on Addgene.org:
- Find plasmids encoding recombinant antibodies at Addgene





Leave a Comment