Thousands of mutations have been implicated in cancer. Yet, the majority of them remain uncharacterized. This represents a considerable barrier to furthering research and developing effective treatments for the disease. Recently, Kenneth Scott’s lab at Baylor College of Medicine and Gordon Mills’s lab at the MD Anderson Cancer Center tackled this problem by developing a moderate throughput functional genomic platform to annotate genetic perturbations that may play significant roles in cancer pathogenesis.
Their work culminated in the first functional characterization of over 1,000 mutations, and the group has deposited several hundred plasmids from this study with Addgene. Read on to learn more about their work, and how these plasmids can help cancer researchers studying an assortment of genes.
Using patient data to identify mutations of unknown importance in cancer
Previously available methods for investigating and functionally validating the thousands of mutations that might play a role in the progression of cancer are often time consuming and expensive, especially when done in vivo. There are also sensitivity issues when attempting to characterize mutations in a pooled cell population, as mutations with strong phenotypic effects can mask weaker ones.
Thus, the need for a faster and higher-throughput in vitro approach. Using data from The Cancer Genome Atlas (TCGA), a catalog of genetic mutations responsible for cancer from genomics data, and large-scale patient cohort data from the MD Anderson Cancer Center patient databases, this research team identified 1,049 perturbations in several genes spanning 33 cancer types. The types of mutations they identified included gene amplifications and fusions, missense mutations, and indels (Ng et al., 2018).
From this data, they generated lentiviral constructs using the High-Throughput Mutagenesis and Molecular Barcoding (HiTMMoB) technique (Tsang et al., 2016), creating a collection of plasmids each carrying a single gene with a defined mutation or gene fusion. Plasmids carrying the corresponding wild-type genes were constructed as well. They called this library the CTD2 FASMIC Plasmid Collection for “Functional Annotation of Somatic Mutations in Cancer.” Of all the mutations and fusions represented in this collection, approximately 80% (over 800) of them had not yet been functionally validated.
In vitro assay allows first-time functional annotation of thousands of mutations
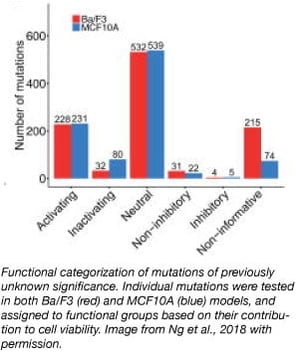 The researchers used the collection to screen individual perturbations in vitro using Ba/F3 and MCF10A cellular models. Ba/F3 and MCF10A are growth factor dependent cell lines and any mutations that that confer proliferation advantages will be revealed in the absence of growth factor. The team assigned the mutations to functional groups, based on how each one impacted cell viability (see figure). Most of the mutations were found to be “neutral,” having no significant effect on cell viability compared to a wild-type control. Approximately ~22% of the mutations, however, significantly increased cell viability, and these “activators” may be the direct drivers of tumor formation. Being able to parse out “neutral” passenger mutations from the “active” drivers is important for the development of targeted therapeutic interventions for cancer.
The researchers used the collection to screen individual perturbations in vitro using Ba/F3 and MCF10A cellular models. Ba/F3 and MCF10A are growth factor dependent cell lines and any mutations that that confer proliferation advantages will be revealed in the absence of growth factor. The team assigned the mutations to functional groups, based on how each one impacted cell viability (see figure). Most of the mutations were found to be “neutral,” having no significant effect on cell viability compared to a wild-type control. Approximately ~22% of the mutations, however, significantly increased cell viability, and these “activators” may be the direct drivers of tumor formation. Being able to parse out “neutral” passenger mutations from the “active” drivers is important for the development of targeted therapeutic interventions for cancer.
This screening platform is an incredible boost to the cancer research community, but what else are these plasmids useful for? They could be used to more thoroughly investigate any of the dozens of wild-type genes and their associated mutants in other model systems. It is possible that a given mutation will have a different phenotypic effect in a different system.
These plasmids can also help scientists understand how a given gene is involved in a complex pathway. Some of the most heavily-studied genes in cancer research, like PIK3CA (Plasmid 116771: pHAGE-PIK3CA) and EGFR (Plasmid 116731: pHAGE-EGFR), are represented in this collection along with hundreds of less well-studied candidates. The possibilities are vast.

References
Ng, Patrick Kwok-Shing, et al. "Systematic functional annotation of somatic mutations in cancer." Cancer Cell 33.3 (2018): 450-462. PubMed PMID: 29533785. PubMed Central PMCID: PMC5926201.
Tsang, Yiu Huen, et al. "Functional annotation of rare gene aberration drivers of pancreatic cancer." Nature communications 7 (2016): 10500. PubMed PMID: 26806015. PubMed Central PMCID: PMC4737758.
Additional resources on the Addgene blog
- Learn about our cancer collection pages
- Read all cancer blog posts
Resources on Addgene.org
Topics: Cancer, Other Plasmid Tools, Plasmids





Leave a Comment