As with DNA isolation, scientists commonly rely on RNA isolation kits to make their life easier. Recently, we published a blog on DNA purification without a kit that outlined several reasons why doing something without a kit has advantages: less plastic waste, less expense, and less of being left with a bunch of random solutions when all the spin columns run out. In this article, we cover the basics of isolating RNA without a kit.
Tips for working with RNA (whether you are using a kit or not)
Although it goes without saying that one should take care whenever doing any type of DNA or RNA purification to avoid contamination, take extra care when performing RNA extraction. RNA is inherently not as stable as DNA- it is single stranded and its ribose groups are susceptible to hydrolysis and heat degradation. Furthermore, RNases, or enzymes that degrade RNA, are especially hardy proteins that are found in and on everything, including your skin. Here are some general tips for working with RNA, even if you are using a kit:
- Always wear gloves, as the RNases on your hands can degrade RNA.
- Keep a clean work area, which may include spraying your bench down with a product to get rid of RNases such as RNaseZAP.
- When harvesting tissues, cells, plants, fungi, or bacteria, keep samples cold and work quickly to mitigate RNA degradation.
- Make sure to use DEPC-treated or RNAse free water. If using DEPC-treated water, autoclave the water to inactivate the DEPC.
- Ensure that plasticware or glassware used is RNase free. RNase-free plasticware is readily available from scientific suppliers and glassware should be treated with a DEPC solution for 1 hour, and autoclaved to remove residual DEPC. Alternatively, glassware can be baked at 180°C for at least 4 hours.
- If your final RNA sample(s) are resuspended in water or TE buffer, store them in a -80°C freezer to prevent RNA degradation. They will degrade in a -20°C freezer.
RNA extraction methods evolved into a simple protocol still used today
There are many alternative methods for isolating DNA without a kit. However, that isn’t the case for RNA extraction and purification. There is one simple method that works, and variations to that method. A major hurdle to developing protocols to isolate RNA was that RNases are commonly found in cells, and without something to block RNase activity upon cell lysis, RNA is degraded. To effectively isolate intact RNA, a fast, strong protein denaturant would be required- something that broke down RNases before RNases had a chance to break down RNA upon cell lysis.
In the late 1970’s, Chirgwin and colleagues showed that a strong protein denaturant, guanidinium thiocyanate, did just this (Chirgwin et al., 1979). They developed a protocol meant for isolating RNA from rat spleens in which they homogenized spleens in a guanidinium thiocyanate solution and spun down the homogenate to remove the insoluble material. Then, the homogenate was loaded onto cesium-chloride gradients and ultracentrifuged for up to 20 hours to separate the intact RNA from DNA and proteins. Although very effective at isolating total RNA, this method requires a lot of time and depending on how many samples you may have, access to one or more large, expensive ultracentrifuge.
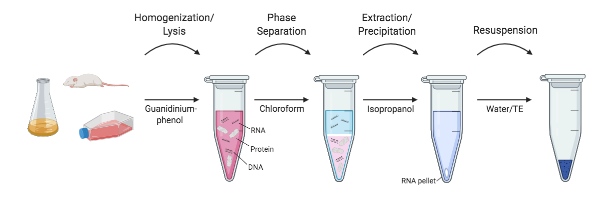 |
| Figure 1: An outline of the different steps in RNA extraction. |
Researchers at the NIH in the mid 1980’s set out to develop a protocol that skipped the ultracentrifugation altogether. Chomczynski and Sacchi showed that RNA could be effectively separated from DNA and proteins by a simple extraction protocol with guanidinium thiocyanate-phenol-chloroform. In this method, samples are still homogenized and lysed in a guanidinium thiocyanate solution. However, instead of RNA separation using cesium-chloride gradients, water-saturated phenol, sodium acetate, and chloroform are added to the homogenate and shaken. After a quick centrifugation (not ultracentrifugation!), the layers of phenol and chloroform separate, and RNA is retained in the top, aqueous layer, while DNA and other proteins are retained in the interphase and bottom, organic layer. The top aqueous layer is extracted and RNA can then be isopropanol precipitated. This method reduced the time it took to isolate RNA from 20+ hours to around 4 hours, and variations on this no-kit method are still widely used today (Chomcynski and Sacchi, 2006).
View our protocol for RNA extraction!
Making the simple protocol even more foolproof (still without a kit!)
As mentioned above, working with RNA requires keeping your samples cold until homogenization and cell lysis. This can be challenging depending on your laboratory situation or tissue collection method, so biotechnology companies have marketed several products that help to further streamline this process and/or stabilize RNA during tissue collection and homogenization. The most widely known of these products is TRIzol® (also called TRI Reagent®, RNAzol®, QIAzol® and sold by many different companies). TRIzol® is an all-in-one acid-guanidinium-phenol solution that combines the homogenization solution and phenol addition of the original no-kit protocol into one step. After homogenization in TRIzol®, insoluble material is removed via centrifugation and the supernatant is extracted with chloroform as in the above no-kit method.
Researchers have also developed ways to “stabilize” RNA within tissues before cell lysis. These products, namely RNAlater® from Thermo and RNAProtect® from Qiagen, are ammonium sulfate-based solutions that work by inhibiting RNase activity within cells or tissues- they don’t actually chemically stabilize the RNA molecules (Allewell and Sarma, 1974). Additionally, ThermoFisher provides a protocol on how to integrate RNAlater® with use of TRIzol® and ammonium sulfate stabilizing solutions can be made in house.
A common problem with no-kit RNA extraction methods is the carryover of DNA that can potentially complicate results of a downstream application such as quantitative PCR to assess gene expression. There are several things that researchers can do to combat this issue. First and foremost, be mindful of your extractions- if you need really clean RNA, it’s important to make sure that when extracting, to only take the aqueous layer to avoid carryover of DNA from the bottom, organic layer. Another trick is to precipitate the RNA using lithium chloride. LiCl solutions selectively precipitate RNA, but not DNA and proteins. Lastly, using a DNase (there are several DNase enzyme products on the market to choose from) on your resuspended RNA sample will help ensure DNA contamination is not an issue.

References
Allewell NM, Sama A (1974) The effect of ammonium sulfate on the activity of ribonuclease A. Biochimica et Biophysica Acta (BBA) - Enzymology 341:484–488. https://doi.org/10.1016/0005-2744(74)90240-x
Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ (1979) Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18:5294–5299. https://doi.org/10.1021/bi00591a005
Chomczynski P, Sacchi N (1987) Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate–Phenol–Chloroform Extraction. Analytical Biochemistry 162:156–159. https://doi.org/10.1016/0003-2697(87)90021-2
Chomczynski P, Sacchi N (2006) The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: twenty-something years on. Nature Protocols 1:581–585. https://doi.org/10.1038/nprot.2006.83
Additional resources on the Addgene blog
- Read blog posts related to plasmid protocols
- Find protocols for purifying DNA without a kit
Resources on Addgene.org
- Check out our molecular biology protocols
- Find protocol videos on our website
- Read our Molecular Biology Guide





Leave a Comment