What are Acoustic Reporter Genes (ARGs)?
Fluorescent and bioluminescent reporter genes are widely used to study gene expression in cells. But these reporters have limited use in vivo because they are based on light which is very easily scattered by tissue. Ultrasound, on the other hand, is a widely available and inexpensive imaging modality with good tissue penetration depth as well as high spatial and temporal resolution, making it ideal for visualizing gene expression in vivo.
But what ultrasound lacked was reporter genes that could produce ultrasound contrast. That is, until earlier members of our lab - the Shapiro Lab - developed Acoustic Reporter Genes (ARGs), which are sets of genes that, when expressed, enable cells to be visualized under ultrasound imaging. Expression of ARGs results in the production of air-filled protein nanostructures called gas vesicles, which scatter sound waves and thereby produce ultrasound contrast. We (the authors) are currently working on improving and optimizing ARGs, with Marjorie focused on bacterial ARGs and Tom focused on mammalian ARGs.
ARG expression in bacteria
The first-generation bacterial ARGs (bARGs), used in bacteria, expressed poorly at body temperature (37oC) and were too burdensome to express in situ. As a result, they had to be expressed in vitro before the bacteria were injected, limiting their applicability. They also did not scatter ultrasound nonlinearly, making them difficult to distinguish from background tissue unless destructive ultrasound imaging was used.
We set out to develop next-generation bARGs that could scatter ultrasound nonlinearly and be easily expressed heterologously by bacteria inside the body. So we screened gas vesicles gene clusters from a diverse set of gas vesicle-expressing species. The gas vesicle genes from Serratia sp. 39006 produced the highest levels of nonlinear acoustic contrast in E. coli. We deleted an unannotated hypothetical protein, improving expression, and named this gene set bARGSer, for bacterial ARG from Serratia sp. 39006.
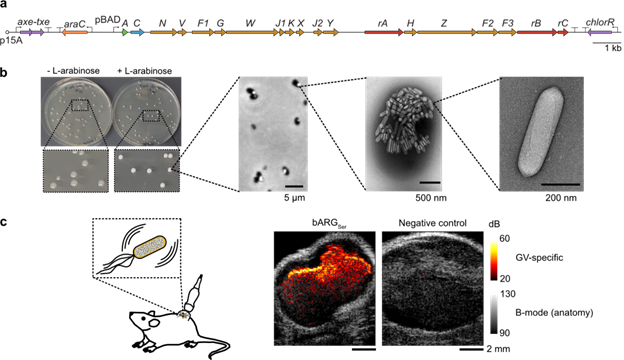
We next tested bARGSer with several inducible expression systems in E. coli. The arabinose-inducible promoter pBAD and a low-copy number origin of replication (p15A) worked best under a wide range of inducer concentrations (0.1-1% L-arabinose) without signs of toxicity or burden relative to a fluorescent protein control. We also added the toxin-antitoxin stability cassette Axe-Txe so that the plasmid could be maintained in the absence of antibiotics, resulting in the plasmid pBAD-bARGSer-AxeTxe (Fig. 1a).
With pBAD-bARGSer-AxeTxe, gas vesicle expression in E. coli is easy: simply grow cells in Lennox LB (10 g/L tryptone, 5 g/L yeast extract, and 5 g/L NaCl) or on LB-agar plates containing glucose and L-arabinose at 37oC for at least 12 hours, or at lower temperatures for longer. Cultures induced with arabinose and colonies on arabinose-containing plates will be visibly white, and gas vesicles in cells are visible under both phase contrast microscopy and electron microscopy (Fig. 1b).
Gas vesicle expression in vivo is also straightforward: after the E. coli have colonized tumors, inject L-arabinose intraperitoneally and wait 24 hours before imaging with ultrasound to observe gas vesicle-specific ultrasound contrast where the bacteria have colonized (Fig. 1c).
|
|
Fig. 1: Expressing bARGSer in E. coli. (a) Plasmid diagram of pBAD-bARGSer-AxeTxe. (b) Methods to assess bARGSer expression, from left to right: colony whiteness on inducer plates, white inclusions in cells with GVs via phase contrast microscopy, GVs visible in cells via TEM, and purified GVs under TEM. (c) Ultrasound imaging of tumor colonization by bARGSer-expressing E. coli. The GV-specific ultrasound signal inside the tumors was overlaid with the anatomical B-mode images in greyscale. |
ARG expression in mammalian cells
The first-generation mammalian ARGs (mARGs) enabled ultrasound visualization of in situ mammalian gene expression in deep tissue. However, not unlike the first-generation bARGs, poor expression and acoustic properties of these gas vesicles made non-destructive specific imaging in tissue difficult. The first-generation mARGs also had a large genetic footprint (two cassettes + a booster cassette), their expression heterogeneity required single cell screening to isolate a clonal cell line capable of producing ultrasound contrast, and gene silencing required addition of chromatic-opening agents to boost expression to appreciable levels.
We wanted to create second-generation mARGs with higher expression, a smaller genetic footprint, and nonlinear acoustic scattering. The best candidate for this was the Anabaena flos-aquae gas vesicle gene cluster because of its small size and its ability to produce gas vesicles with nonlinear contrast.
Increasing expression and decreasing genetic footprint
We took a cue from the native expression patterns of the major structural protein GvpA, which seemed overexpressed compared to accessory proteins. Excess gvpA plasmid added into the transfection mixture in HEK293T cells gave drastic improvement in expression. Stabilizing the gvpA transcript with WPRE reduced the number of copies of gvpA needed to increase expression.
We were able to express Anabaena GVs in mammalian cells without the shell stiffening protein GvpC, which allowed the GVs to be flexible and produce nondestructive nonlinear ultrasound contrast. It turned out that Anabaena mARGs did not require a booster cassette, either, allowing us to reduce the genetic footprint yet again.
This also led to improved expression homogeneity, as the cells could be batch-sorted to isolate a polyclonal cell line with strong gas vesicle expression and robustness without chromatin-opening agents. It ended up being very straightforward to engineer various cell lines from different tissues and mammalian species to produce gas vesicles.
New mARG plasmids
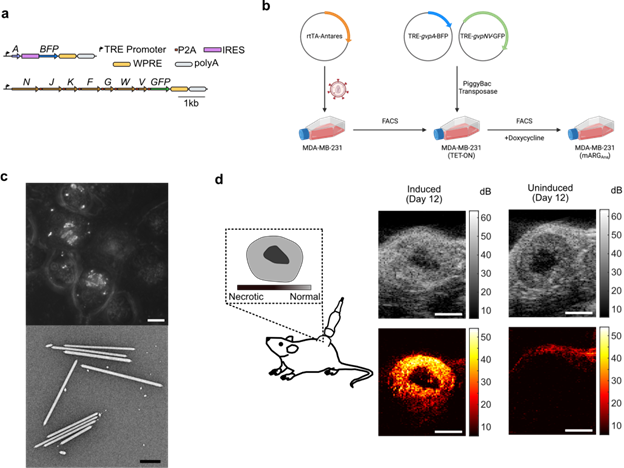
Our new mARGs come as a set of two PiggyBac integratable cassettes, both with doxycycline inducible promoters (Fig. 2a). The first cassette contains the major structural gene gvpA marked with EBFP2 and the second cassette contains the P2A-linked accessory genes gvpNJKFGWV marked with Emerald GFP. These plasmids can be used directly in transient transfection in rtTA+ cell lines (such as HEK293-TETOn cells), or they can also be integrated into the genome by cotransfecting them with a plasmid expressing PiggyBac transposase. In rtTA-cell lines, it is better to first engineer the cell line with rtTA, then integrate the mARGs using PiggyBac.
Gas vesicles are detectable under ultrasound and phase contrast microscopy just one day after induction of expression… but longer expression leads to greater amount of gas vesicles and increased ultrasound contrast. In all the cell lines we tested, 1 µg/mL doxycycline was sufficient to saturate expression.
When engineering a cell line, cotransfect the mARG cassettes 2:1 (A:N-V) with PiggyBac transposase and outgrow the cells for a several generations to dilute the unintegrated plasmids (Fig. 2b). Next, perform a short (12-hour) induction with doxycycline prior to FACS, gating for the top quartile (or higher) of BFP and GFP producing cells. After FACS, return the cells to doxycycline-free media and expand the cells. Use a subset of the cells to verify the mARG positivity, which can be observed under phase contrast microscopy and TEM (Fig. 2c), and ultrasound imaging. If needed, resort the cells again for improved purity and contrast. For in vivo experiments, simply induce injected engineered cells via regular intraperitoneal doxycycline injections to enable their tracking with ultrasound imaging (Fig. 2d).
Creating a cell line can be a time-intensive endeavor. That is why we also included an analogous set of transient constitutively-expressed mARG plasmids driven by CMV promoters. Labs that are new to the mARG expression are advised to first try to produce GVs in their own lab using the most reliable method, which is the transient cotransfection of the mARG plamsids into HEK293T cells using a chemical transfection reagent (e.g. PEI-MAX). Simply cotransfect a mixture of 1248 ng of CMV-gvpA-IRES-BFP plasmid and 954 ng CMV-gvpNJKFGWV plasmid into 70%-confluent well of HEK cells in a 6-well dish and change the media every day for three days. GVs should be visible under phase contrast starting day 2 after transfection.
|
| Fig: 2: Expressing mARGAna in MDA-MB-231 cells. (a) Plasmid diagram of mARGAna. (b) Schematic of the general method to create your own mARGAna cell line. (c) Z-projected phase contrast images of GV expressing MDA-MB-231 cells (top, scalebar = 10µm) and TEM of GVs purified from MDA-MB-231 cell lysate after 4-days of expression (bottom, scalebar = 0.5µm). (d) Ultrasound imaging of MDA-MB-231 tumors after 12-days of in situ expression. The GV-specific ultrasound signal is in hot and the anatomical B-mode images are in greyscale. (scalebar = 2mm) |
We hope you enjoy playing with our genes. Have fun!


 About the authors: Marjorie Buss is a PhD student studying chemical engineering. Mengtong "Tom" Duan is a PhD student studying bioengineering. They are both members of the Shapiro lab at Caltech.
About the authors: Marjorie Buss is a PhD student studying chemical engineering. Mengtong "Tom" Duan is a PhD student studying bioengineering. They are both members of the Shapiro lab at Caltech.
References and Resources
References
Piraner, D. I. et al. Going Deeper: Biomolecular Tools for Acoustic and Magnetic Imaging and Control of Cellular Function. Biochemistry 56, 5202–5209 (2017).
Bourdeau, R. W. et al. Acoustic reporter genes for noninvasive imaging of microorganisms in mammalian hosts. Nature 553, 86–90 (2018).
Farhadi, A., Ho, G. H., Sawyer, D. P., Bourdeau, R. W. & Shapiro, M. G. Ultrasound imaging of gene expression in mammalian cells. Science 365, 1469–1475 (2019).
Maresca, D. et al. Nonlinear ultrasound imaging of nanoscale acoustic biomolecules. Appl. Phys. Lett. 110, 073704 (2017).
Hurt, R. C. et al. Genomically mined acoustic reporter genes for real-time in vivo monitoring of tumors and tumor-homing bacteria. Nat Biotechnol 1–13 (2023) doi:10.1038/s41587-022-01581-y
Topics: Other Plasmid Tools, Plasmids





Leave a Comment