Biomedical research has a long and well-documented history of racial bias, which still impacts and influences current research. Understanding the depth and breadth of this bias, and then using that understanding to intentionally remove racial bias from work being done today, is a nearly entirely separate effort from acknowledging its existence.
Studying ancestral group dependencies
Take, for instance, a recent preprint released by the Boehm and Beroukhim labs at the Broad Institute of MIT and Harvard, Dana-Farber Cancer Institute, and Koch Institute, where an exploration into the effects of ancestry and germline sequence variation in cancer cell lines took them down unexpected paths. Though this research uses ancestry groups rather than self-reported race, it highlights the myriad issues researchers need to reflect upon before they attempt to make essentialist connections between biology and ancestral groups or, by extension, race.
Many researchers do large-scale CRISPR screens in cancer cell lines, looking to uncover genetic dependencies of individual tumors, which can lead to new cancer targets and related biomarkers . Data from such screens are often uploaded to the Cancer Dependency Map (DepMap), a cancer dependency database used to make this information accessible to the research community. Sean Misek and Aaron Fultineer in the Beroukhim and Boehm labs pulled data from more than 1000 CRISPR screens in DepMap to see if they could identify the ancestry of the cell lines (European, East Asian, Indigenous American, Africa, or South Asian) used in the screens based on the cells’ germline genetic variations. They then wanted to identify cancer-associated genetic dependencies associated with each ancestry group.
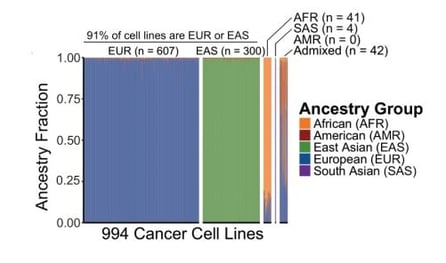
Since about 90% of the cell lines used in DepMap CRISPR screens are of either European or East Asian ancestry, Sean and Aaron knew that their study had significant statistical limitations, and expected to find only a few, very strong associations.
Unexpected results
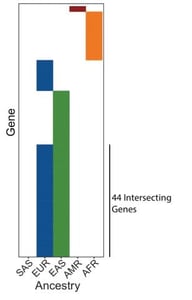
Their results showed 96 strongly correlated genetic dependencies that were clearly associated with European or East Asian ancestry groups, far more than expected. Like any diligent scientist, they sought to understand these strange results by digging deeply into their data.
|
| Fig. 1: Genomic fraction for each of five major ancestry groups for 994 of the Cancer Dependency Map cancer cell lines (left). Heatmap indicating the breakdown of ancestry associated dependencies across ancestry groups (right). |
Seeking an explanation
First, they looked for a biological explanation, a relationship between the genetic dependencies they identified and their associated ancestries. While there were some exciting relationships identified, 48% were found to be artificial, so that didn't seem a plausible explanation. Next, they hypothesized that differences in gene expression could explain why some ancestry groups were differentially dependent on certain genes. This didn’t prove to be correct, either.
However, when they analyzed the variations in gene expression, they found that for approximately 40% of the identified genes, the genes were not expressed at all. This unexpected discovery, which didn’t make sense in any kind of biological framework, suggested to them that their findings may be the result of a technical artifact that introduced an ancestry bias into their experiments instead of a true correlation.
It turned out there was indeed a bias, and it was coming through an unexpected source: the CRISPR guides, short pieces of RNA that bind to a cell’s DNA, telling the Cas machinery where to cut.
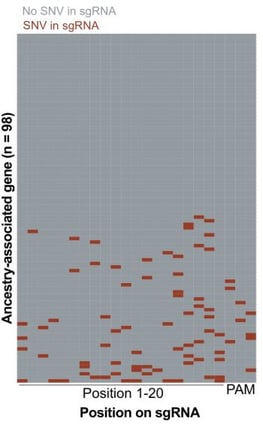 |
| Fig. 2: Heatmap indicating loci where SNPs reside on sgRNA target sequences for sgRNAs that target ancestry-associated genes. |
What does the reference genome actually reference?
The CRISPR guides used in the screens they analyzed were designed to have a perfect match to the reference genome – even single mismatches within a CRISPR guide can prevent the guide from binding to the genome or prevent Cas9 from cutting, both of which can introduce significant bias into the results. But the reference genome does not represent the diversity of genetic variation across the human population. Rather, it primarily reflects European ancestry.
Understanding the impact
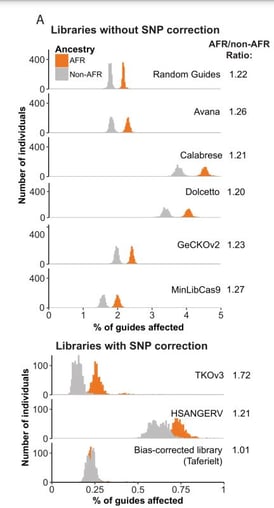 |
| Fig. 3: Histograms indicating the frequency (x-axis) which SNPs map to the targeting sequences of guides across 8 CRISPR libraries. Samples are divided into those of African ancestry (orange) and those of other ancestry groups (grey). |
Misek and Fultineer analyzed the gnomAD dataset, which captures a broader swath of human genetic diversity, to better understand how the use of a primarily European reference genome could affect their analysis. Their work revealed that cell lines from individuals of African descent have approximately 20% more single nucleotide polymorphisms (SNPs) that map to guides in the Broad’s Genomic Perturbation Platform Avana library (used by the Cancer Dependency Map) than lines from European or East Asian ancestries. These SNPs, however, were not accounted for in the CRISPR guides they used, leading to mismatches between the guides and the lines’ genomes, and a reduction in the number of the dependencies detected.
Given this new information, Misek notes that in theory, if the DepMap analysis had an equal representation of every ancestry group, they should have found more ancestry-associated dependencies in African cell lines than in any other ancestry group. With the existing methodology, they estimate that the African ancestry-associated cancer dependencies are undercounted by approximately 20%. In some cases, this may lead researchers to deprioritize genes that could be good targets for therapeutic development, particularly in cancers that are more common in individuals of African descent.
Correcting the bias
While their thorough analysis uncovered some concerning biases within widely used CRISPR screens, they had also found the information they needed to begin correcting the issue. Working with James McFarland, Jeremie Kalfon, Javad Noorbakhsh, Isabella Boyle, and Joshua Dempster from The Cancer Data Science team at Broad, they were able to change their analysis to use a corrected genome map. Now, any data that came from CRISPR guides mapped to a SNP would be ignored. Working with Lia Petronio and Katherine Huang from Broad’s Pattern team, they designed a tool that could analyze CRISPR sgRNAs and report the likelihood of any given sgRNA having an ancestral bias. Next, they worked with John Doench, David Root, Tom Green, and Adam Brown from Broad's Genetic Perturbation Platform to design a new library that is agnostic to ancestry which will reduce the likelihood of generating genetic dependency data with an ancestry-based bias.
Moving forward
It took a five-team response to address this issue, but it still points to a far larger and more complex problem endemic to biomedical research: pervasive bias that is easy for researchers to ignore or miss. The lack of representation in commonly used cell lines led to an incorrect assumption of what a “default” genome looks like, which led to the incorporation of ancestral bias into genome-wide screens, introducing bias into all work based on these and any other CRISPR screens using the same reference genomes and libraries. Further complicating the issue is the long and ugly history of reporting false essentialist links between biology and ancestry group as a means of perpetuating racial bias. It is easy, even unintentionally, to attribute to an ancestry group what is actually an artifact introduced by bias within the design of the experiment or dataset itself.
It is important for scientists to not only be aware of the potential for bias but to actively look for and consider bias as a confounding factor when interpreting findings, particularly when researching ancestry groups. And, of course, it’s imperative for scientists to be aware of and actively use tools designed to help reduce or eliminate bias, just like the ones made available through the work done in this preprint.
Resources and references
References
Resources on ancestral bias and the human genome
Wang, T., Antonacci-Fulton, L., Howe, K. et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446 (2022). https://doi.org/10.1038/s41586-022-04601-8
Lewis, Anna CF, Santiago J. Molina, Paul S. Appelbaum, Bege Dauda, Anna Di Rienzo, Agustin Fuentes, Stephanie M. Fullerton et al. Getting genetic ancestry right for science and society." Science 376, (2022): 250-252.
Carlson J, Henn BM, Al-Hindi DR, Ramachandran S. Counter the weaponization of genetics research by extremists. Nature 610, 444-447 (2022). doi: 10.1038/d41586-022-03252-z.





Leave a Comment