The Rett Syndrome Research Trust (RSRT) has an ambitious goal of curing Rett Syndrome and Addgene is thrilled to partner with them to launch a Rett Syndrome Plasmid Collection and Resource Center for Rett Syndrome research. With a shared passion for facilitating research and discovery, Addgene and RSRT have consolidated and centralized a collection of molecular biology tools so that both veteran and new Rett researchers can find the reagents and resources they need to cure Rett Syndrome.
If you’re new to Rett Syndrome, this blog post describes the history of Rett Syndrome research, the role of the MECP2 gene, and highlights some of the latest breakthrough therapeutic approaches.
 What is Rett Syndrome?
What is Rett Syndrome?
Caused by a mutation in the methyl-CpG binding protein 2 (MECP2) gene, Rett Syndrome is a rare, progressive, neurodevelopmental genetic disorder that affects primarily girls. After a period of seemingly normal development through ages 6 to 18 months, individuals with Rett syndrome experience regression of previously acquired skills such as speech, coordination, purposeful use of hands, and walking. They may exhibit slow head growth, develop seizures, cardio-respiratory dysfunction, distinctive hand movements, and show autistic-like behaviors.
The role of MECP2 in Rett Syndrome
Rett Syndrome was first described in 1966 by Austrian physician, Andreas Rett, who tried to raise awareness and published his observations in a German medical journal. Decades later and after years of painstaking work trying to isolate the causative gene for Rett Syndrome, Huda Zoghbi's lab discovered the first disease-causing mutations in the MECP2 gene leading to Rett Syndrome (Amir et al., 1999). We now know that MECP2 binds to methylated CpG dinucleotides and CAC trinucleotides during neuronal maturation (Tillotson & Bird, 2019) and is a global transcriptional regulator of thousands of genes. Studies have suggested roles in transcriptional repression, activity dependent de-repression, chromatin remodeling, gene activation, and more. Loss of MECP2 in adult animal models results in rapid symptom onset, suggesting that MECP2 is also essential after development (McGraw et al., 2011).
Adrian Bird’s 2007 landmark paper in Science showed that dramatic reversal of disease symptoms in mice is possible (Guy et al., 2007). They accomplished this using Cre-Lox mouse models in which the endogenous MECP2 gene was disrupted and silenced by a Lox-Stop cassette, resulting in the neurological deficits observed in Rett Syndrome. Restoring the MECP2 gene by excision of the Stop cassette using tamoxifen-inducible Cre not only halted the neurological damage but completely reversed it, regardless of age or symptom severity. This exciting finding injected hope that a similar reversal may be possible for children and adults with Rett Syndrome.
Since these discoveries, scientists have studied how different parts of MECP2 influence Rett Syndrome. Zoghbi’s lab found that a small DNA binding motif in MECP2 interacts with the minor groove of AT-rich DNA sequences to regulate transcription (Baker et al., 2013). Mutations that affect this interaction to varying degrees determine the clinical severity of different MECP2 mutations. More recently, the Bird lab found that only a portion of the MECP2 protein is necessary to rescue the neurological defects of Rett syndrome (Tillotson et al., 2017).
Advancing gene therapies for Rett Syndrome
MECP2 follows the “Goldilocks” principle, in which too much of the MECP2 protein is as detrimental as too little, resulting in MECP2 duplication syndrome or Rett Syndrome, respectively. One of the main challenges facing Rett researchers is how to deliver the precise amount of protein to each brain cell to restore physiological levels. Much research is now focused on therapeutic strategies that use gene replacement therapy, gene editing, MECP2 reactivation, RNA editing, RNA trans-splicing, and protein replacement to reverse the impacts of this disease.
As a non-degenerative, monogenic disease that has shown reversibility of symptoms in animal models, Rett syndrome is a strong translational candidate for gene replacement therapy. In 2014, RSRT brought together a strong collaboration between two gene therapy labs (Brian Kaspar and Steven Gray) and two MECP2 labs (Gail Mandel and Stuart Cobb) to create the Gene Therapy Consortium. Leveraging their combined technical expertise to develop a viable therapeutic, the researchers developed adeno-associated viral (AAV) vectors for the targeted delivery of a functional MECP2 gene in preclinical animal studies with positive results (Sinnett et al., 2017; Powers et al., 2019). Based on the work of the consortium, two gene therapy candidates advanced into clinical development programs in biopharma. AveXis, co-founded by Kaspar, acquired by Novartis and rebranded to Novartis Gene Therapies, and Taysha Gene Therapies, founded on Gray’s work, are each expected to advance their respective gene therapies to clinical trials in 2021.
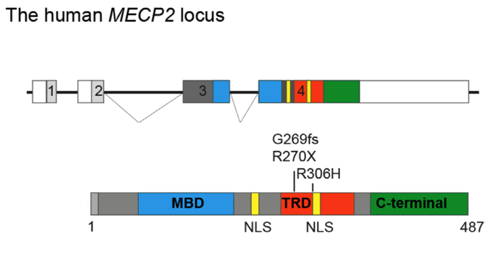 |
| Figure 1: Schematic of the human MECP2 locus and MECP2 protein, showing mutations corrected by CRISPR/Cas9 in wild-type human iPSCs. Image modified from Le et al., 2019. |
Gene editing via CRISPR/Cas9 and RNA-mediated correction of MECP2 mutant alleles, in particular, have shown exciting and promising results for repairing mutations in models of neurological disease. Klaus Rajewsky’s group recently used the CRISPR/Cas9 system to both precisely insert the MECP2-R270X mutation into the MECP2 gene in wild-type human iPSCs and then to successfully repair the mutation (Le et al., 2019). Beam Therapeutics, co-founded by Harvard Investigators and Addgene depositors David Liu, PhD, Keith Joung, PhD, and Feng Zhang, PhD, is pursuing CRISPR/Cas9 technology for precise correction of Rett syndrome mutations.
Gail Mandel and her team have shown for the first time that programmable RNA editing can repair a gene in a living animal across multiple cell types using a mouse model for a human neurological disease (Sinnamon et al., 2020). They introduced a mouse MECP2 guide RNA and a hyperactive version of the human RNA base editing enzyme (“Editase”), ADAR2, into the hippocampus of a Rett Syndrome mouse model using an adeno-associated viral (AAV) vector. The editase repaired half of the mutant MECP2 RNA in each of three neuronal subtypes of the hippocampus. This technology has formed the basis of the Rett syndrome program being pursued at Vico Therapeutics, co-founded by Gail Mandel.
There are numerous technical challenges to overcome for both of these approaches, such as delivery of the editing machinery to the brain, minimizing off-target editing, and, for RNA editing, fine-tuning dosage so that enough wild-type MECP2 is made inside the cells, but this is exciting progress in tackling the root cause of Rett Syndrome in novel ways.
Addgene’s MECP2 Plasmid Collection and Resource Center will continue to expand as Rett scientists deposit their MECP2-related plasmid tools with Addgene and share additional resources. Plasmid tools to facilitate MECP2 curative strategies, such as CRISPR and RNA editing or the induction of iPSCs, are available in the Resource Center. Researchers can also find links to cell line repositories including patient-derived iPSCs, MECP2 animal models, tissue banks, Rett databases, and other helpful tools.

References:
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188 . https://doi.org/10.1038/13810
Amir RE, Zoghbi HY (2000) Rett syndrome: Methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations. Am J Med Genet 97:147–152 . https://doi.org/10.1002/1096-8628(200022)97:2<147::aid-ajmg6>3.0.co;2-o
Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY (2013) An AT-Hook Domain in MeCP2 Determines the Clinical Course of Rett Syndrome and Related Disorders. Cell 152:984–996 . https://doi.org/10.1016/j.cell.2013.01.038
Guy J, Gan J, Selfridge J, Cobb S, Bird A (2007) Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science 315:1143–1147 . https://doi.org/10.1126/science.1138389
Le TTH, Tran NT, Dao TML, Nguyen DD, Do HD, Ha TL, Kühn R, Nguyen TL, Rajewsky K, Chu VT (2019) Efficient and Precise CRISPR/Cas9-Mediated MECP2 Modifications in Human-Induced Pluripotent Stem Cells. Front Genet 10: . https://doi.org/10.3389/fgene.2019.00625
McGraw CM, Samaco RC, Zoghbi HY (2011) Adult Neural Function Requires MeCP2. Science 333:186–186 . https://doi.org/10.1126/science.1206593
Powers S, Miranda C, Dennys-Rivers C, Huffenberger A, Braun L, Rinaldi F, Wein N, Meyer KC, Solano S, Nguyen K , Lang E, Kaspar AA, Foust KD, Thomsen G, Fugere M, Kaspar BK (2019) Rett syndrome gene therapy improves survival and ameliorates behavioral phenotypes in MeCP2 null (S51.002). Neurology 92 (15 Supplement) . http://n.neurology.org/content/92/15_Supplement/S51.002.abstract
Sinnamon JR, Kim SY, Fisk JR, Song Z, Nakai H, Jeng S, McWeeney SK, Mandel G (2020) In Vivo Repair of a Protein Underlying a Neurological Disorder by Programmable RNA Editing. Cell Reports 32:107878 . https://doi.org/10.1016/j.celrep.2020.107878
Sinnett SE, Hector RD, Gadalla KKE, Heindel C, Chen D, Zaric V, Bailey MES, Cobb SR, Gray SJ (2017) Improved MECP2 Gene Therapy Extends the Survival of MeCP2-Null Mice without Apparent Toxicity after Intracisternal Delivery. Molecular Therapy - Methods & Clinical Development 5:106–115 . https://doi.org/10.1016/j.omtm.2017.04.006
Tillotson R, Selfridge J, Koerner MV, Gadalla KKE, Guy J, De Sousa D, Hector RD, Cobb SR, Bird A (2017) Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 550:398–401 . https://doi.org/10.1038/nature24058
Tillotson R, Bird A (2020) The Molecular Basis of MeCP2 Function in the Brain. Journal of Molecular Biology 432:1602–1623 . https://doi.org/10.1016/j.jmb.2019.10.004
Topics: Other





Leave a Comment