
We're excited to announce that, as in years past, we've been working with the Rare Genomics Institute to provide plasmid grants to researchers working on rare diseases through the BeHEARD Award.
Congratulations to Paula Videira, Michael McMurray, and Richard Gronostajski who each are being awarded free plasmids form Addgene to accelerate their research. Read on to learn how these researchers plan to use plasmids from Addgene to advance their research on diseases of glycosylation, arginine mutagenesis, and neural development.
Paula Videira: Congenital disorders of glycosylation
Paula Videira is an Assistant Professor at the Faculty of Sciences of NOVA University in Lisbon, Portugal. She leads the Glycoimmunology group based at the Research Unit on Applied Molecular Biosciences (UCIBIO) focused on understanding the physiological and pathological roles of human cell glycans. Accordingly, she joined efforts with the Portuguese Association for Congenital Disorders of Glycosylation (APCDG) and an international platform for CDG research, named the CDG&Allies – Professionals and Patient Associations International Network (CDG&Allies-PPAIN).
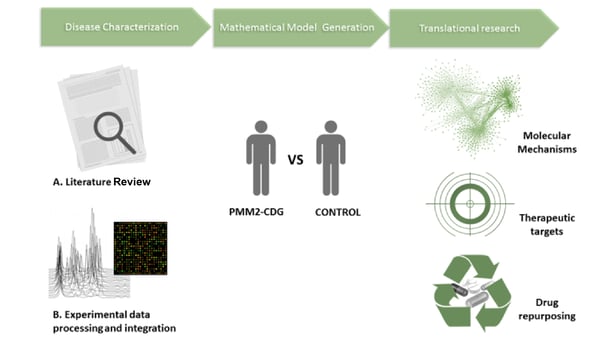
Congenital disorders of glycosylation (CDG) are a group of rare inherited metabolic diseases with severe and life-threatening consequences. They result from defects in different steps in the glycan biosynthetic pathway and have a wide range of clinical manifestations. PMM2-CDG is the most frequent CDG type characterized by deficient phosphomannomutase (PMM) activity due to mutations in the PMM2 gene. Videira’s group recently reviewed and reported that PMM2-CDG patients suffer from immunological defects. These are characterized by a predisposition to severe and recurrent infections along with a mild or exacerbated inflammatory response which leads to nearly 25% child mortality.
Since glycans play an essential role in almost every aspect of immunology, participating in events such as cell development, recognition and migration, the immune compromise observed in CDG patients is not surprising. However, the molecular and cellular mechanisms behind the immune defects in CDG patients are still poorly studied and understood.
In line with this, Prof. Videira and collaborators will use Addgene plasmids to immortalize CDG patient-derived immune cell lines and use these lines to study how immune processes and biological pathways are altered in PMM2-CDG. These studies are expected to identify new therapeutic targets/solutions and enable scientists to discover if known drugs can be used to treat these diseases (drug repositioning). The ultimate goal of this work is to lower the burden of immune symptoms on CDG patients and to increase their quality of life by reducing their risks for infection and other pathological conditions.
References:
Monticelli, M., Ferro, T., Jaeken, J., dos Reis Ferreira, V. & Videira, P. A. Immunological aspects of congenital disorders of glycosylation (CDG): a review. J. Inherit. Metab. Dis. 39, 765–780 (2016). PubMed PMID: 27393411.
Michael McMurray: Rescuing arginine mutations in rare disease with guanidine
Michael McMurray is an Associate Professor in the Department of Cell and Developmental Biology at the University of Colorado Anschutz Medical Campus in Aurora, Colorado, USA. The primary focus of his lab’s research is the mechanisms by which proteins assemble into functional multi-subunit macromolecular complexes, but a serendipitous discovery led them to investigate the use of a common laboratory chemical, guanidine hydrochloride, as a potential treatment for a large number of rare genetic diseases.
Spontaneous mutations are a major cause of rare diseases. Spontaneous mutations that change protein sequence affect arginine (Arg) amino acids more frequently than any other. Indeed, nearly 20% of the single missense mutant alleles that cause human disease change Arg to another amino acid. Clearly, the development of a pharmacological intervention that rescues the function of even a small fraction of Arg-mutant proteins could improve health outcomes for millions of individuals worldwide.
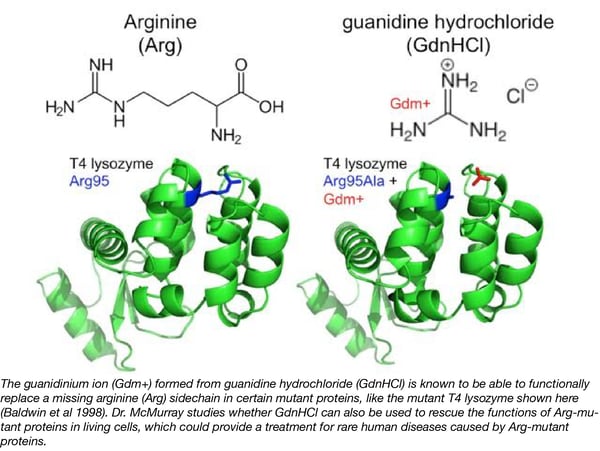
Arg is unique among amino acids in that it possesses a guanidine moiety at the end of its side chain. Decades ago, other labs discovered that the in vitro functions of 11 different Arg-mutant enzymes could be rescued by supplementation with guanidine hydrochloride (GdnHCl). Rescue occurs through guanidinium cation (Gdm+) binding in the location formerly occupied by the Arg sidechain. Although not related to this rescue function, GdnHCl was approved by the FDA in 1939 for the treatment of botulism and Lambert-Eaton myasthenic syndrome (LEMS) because, at high concentrations (25 mM), Gdm+ binds to and inhibits a potassium channel, causing release of acetylcholine. Given its unique molecular biology and previous approval by the FDA, GdnHCl could, in principle, provide an effective treatment for rare diseases caused by Arg-mutant proteins, but there is currently no evidence of Arg-mutant protein rescue in living cells.
The McMurray lab found that low concentrations (≤3 mM) of GdnHCl can functionally rescue multiple Arg-mutant proteins in living baker’s yeast cells, including Arg-mutant yeast homologs of human proteins that cause rare diseases. They now plan to test a large number of other disease-linked Arg-mutant proteins both in yeast and human cells in order to determine the specific molecular features that allow such “chemical rescue” in vivo. Engineering the human disease alleles into cells for testing in the lab will be greatly facilitated by CRISPR reagents and other specific gene sequences available from Addgene. Dr. McMurray hopes that by demonstrating GdnHCl chemical rescue of a variety of disease-linked Arg-mutant proteins in the lab, clinicians might consider prescribing GdnHCl to patients suffering from rare diseases arising from Arg mutations.
References:
Baldwin, Enoch, et al. "Generation of ligand binding sites in T4 lysozyme by deficiency-creating substitutions." Journal of molecular biology 277.2 (1998): 467-485. PubMed PMID: 9514755.
Richard Gronostajski: NFI-related diseases of neural development
Richard Gronostajski is a professor at the University of Buffalo. His research focuses on understanding the structural determinants of protein-DNA interactions and how these interactions regulation gene expression.
Recent studies have shown that the conditions Sotos-type 2 and Marshall Smith Syndrome are due to mutations in the NFIX gene. These diseases are characterized by overgrowth phenotypes and neural developmental delay. Mutations in NFIA and NFIB can also cause developmental delay in patients. We have been studying the function of the mouse Nfia, Nfib and Nfix genes in brain development since 2000 and have shown several mechanisms by which they affect brain and muscle development. The Gronostajski lab has generated mice wherein it's possible to delete the functional portion of the Nfix gene using Cre recombinase and to generate Nfix-negative neural stem cells. The lab will use plasmids from Addgene to construct NFIX expression vectors as well as reporters of NFIX function. They will then mutagenize the NFIX gene and test whether variants of unknown significance (VUSs) that are frequently found in genetic testing for syndromes that include developmental delay, result in loss of function or dominant negative NFIX phenotypes.
Developing a rapid test for the functional activity of VUSs will enable more accurate diagnosis of NFIX-related syndromes (Sotos-type 2 and Marshall-Smith) and could be expanded to NFIA-, NFIB- and NFIC-related human syndromes. As therapies are developed for specific NFI-related genetic disorders, these will also allow better treatment of these related syndromes.
Many thanks and best of luck to the 2018 BeHeard Award winners!
.jpg?width=100&name=Foto%20Paula%20(1).jpg) Paula Videira is the team leader of the Glycoimmunology group at UCIBIO, FCT-UNL, Portugal. Her main work focuses on unraveling the pathophysiological role of glycans. Follow her on Facebook: @glycoimmunologygroup.
Paula Videira is the team leader of the Glycoimmunology group at UCIBIO, FCT-UNL, Portugal. Her main work focuses on unraveling the pathophysiological role of glycans. Follow her on Facebook: @glycoimmunologygroup.
 Michael McMurray is an Associate Professor in the Department of Cell and Developmental Biology at the University of Colorado Anschutz Medical Campus in Aurora, Colorado, USA.
Michael McMurray is an Associate Professor in the Department of Cell and Developmental Biology at the University of Colorado Anschutz Medical Campus in Aurora, Colorado, USA.
 Richard Gronostajski is a professor at the University of Buffalo. His research focuses on understanding the structural determinants of protein-DNA interactions and how these interactions regulation gene expression.
Richard Gronostajski is a professor at the University of Buffalo. His research focuses on understanding the structural determinants of protein-DNA interactions and how these interactions regulation gene expression.

Topics: Addgene News, Awards





Leave a Comment