By Multiple Authors
Read More
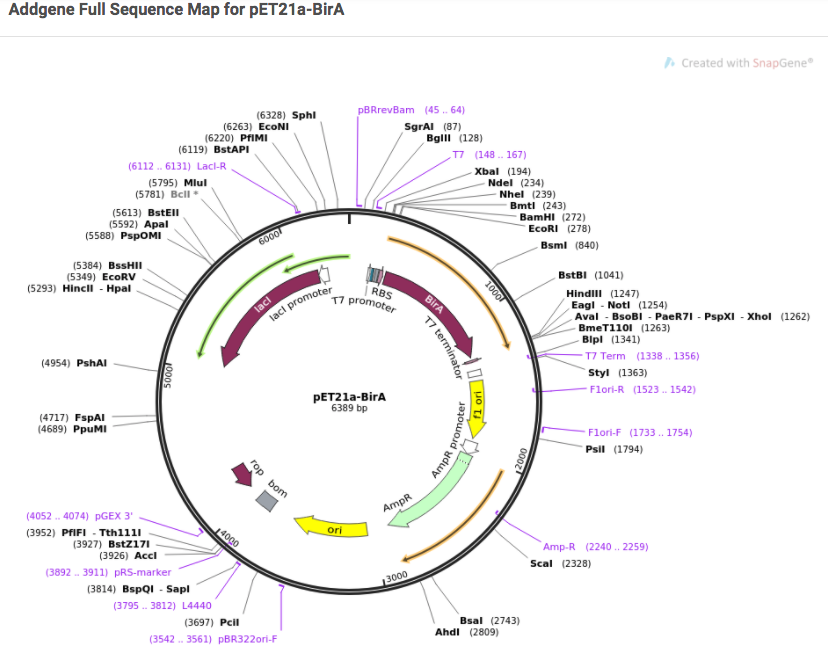
Primer design. Plasmid mapping. DNA sequence analysis. We all have our favorite tools for tackling these particular tasks, but they tend to be scattered about the internet. To help you keep your virtual molecular biology toolbox organized, today’s post features a list of free ...
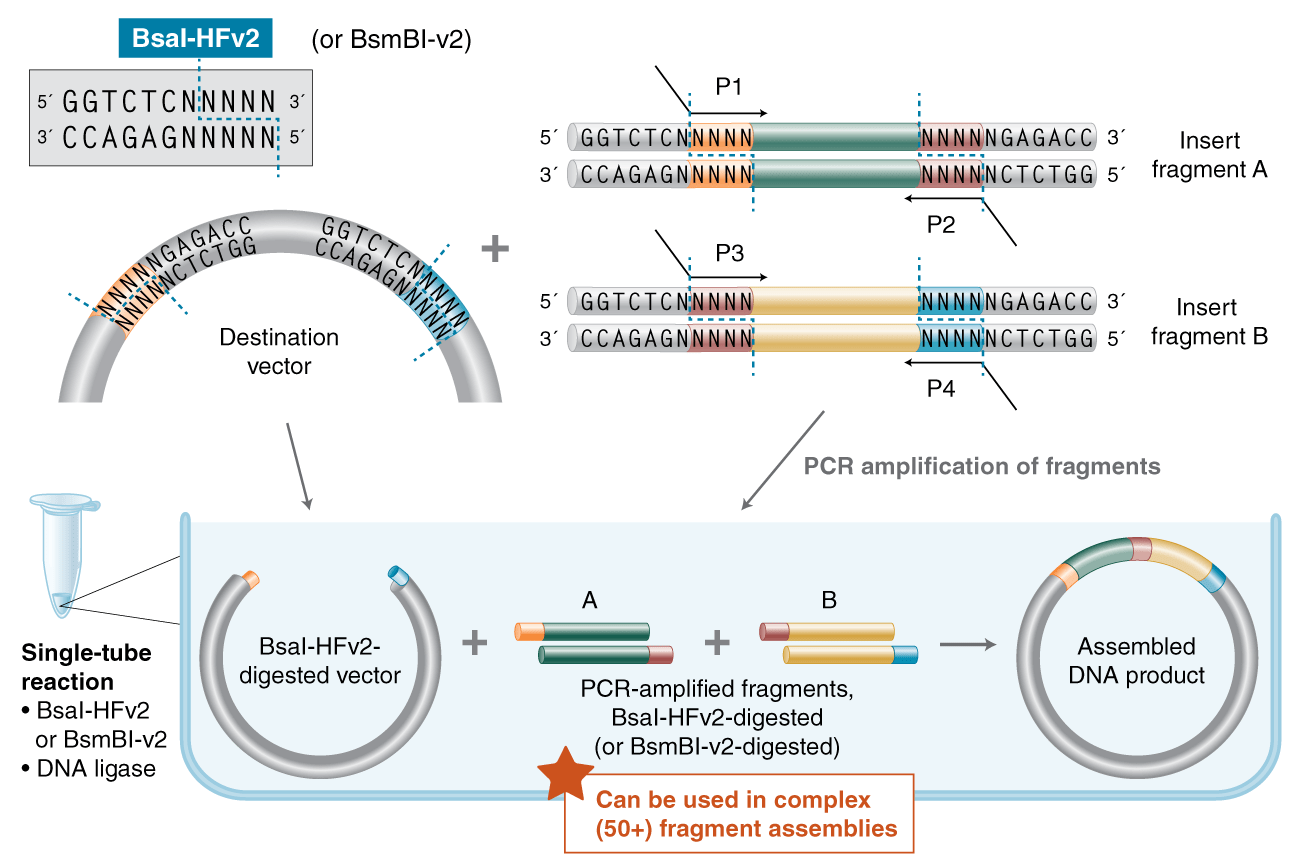
If you’ve ever used Golden Gate Assembly for cloning, you might be familiar with the rules of thumb for designing your overhang sets. But are those rules the best way to design GGA overhang sets, particularly for high-complexity reactions?
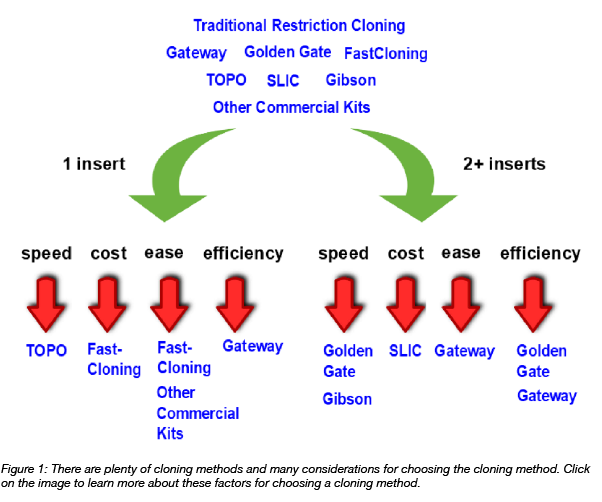
This post was originally written by Jennifer Tsang and updated by Rachel Leeson. Have you ever wondered how long it takes to make a plasmid? Or how much time you have to spend cloning before you can start your experiment? What about all the reagents you need to order? Sometimes, ...
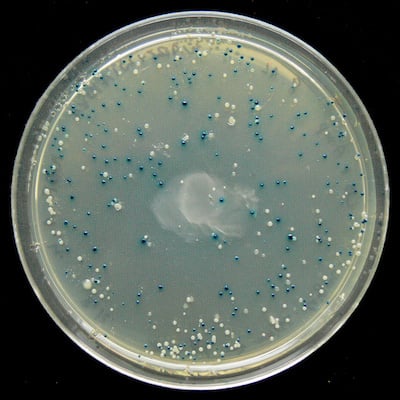
If you’re cloning a plasmid, you’ll need a way to find the needle in the haystack: the one perfect clone that contains the plasmid you’re looking for out of the many cells that don’t. One way to begin the search is by using selection strategies, where only cells that have gained ...
What if after ordering a plasmid you didn't have to grow the bacteria and prep the plasmid before you begin your cloning experiment? What if after receiving the plasmid from Addgene you could directly start your digest, PCR, or transformation? Great news. A subset of our ...
Not all plasmid preps are the same. Before purifying a plasmid from a bacterial culture, it is important to consider your experiment. It will dictate the amount of DNA you need, and at which level of purity. Based on these premises we can classify a plasmid preparation in 3 ...
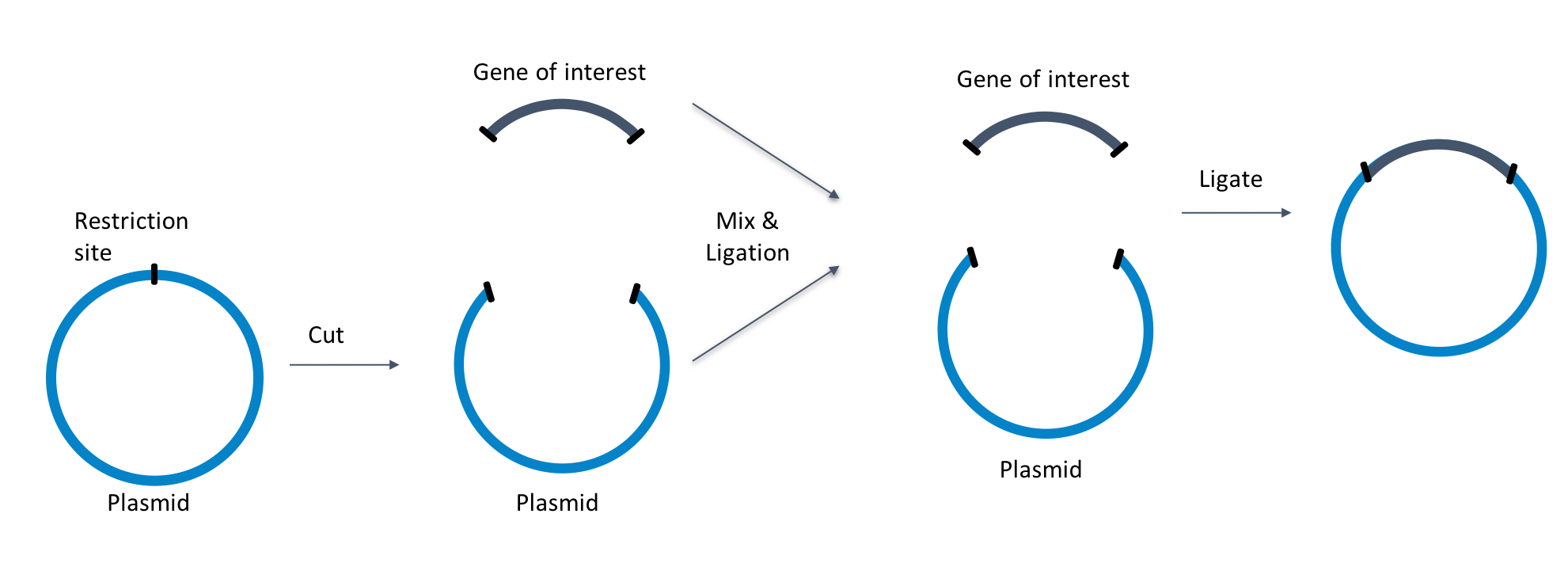
This post was contributed by Oskar Laur, head of the custom cloning core at Emory University, and Paolo Colombi, a product development scientist at Addgene. Cloning can be quite an arduous process. The PCR could fail to produce a product, the transformation may not result in any ...