This post was originally written by Melina Fan and updated Nov 3, 2022 by Susanna Stroik.
You’ve designed your gRNA and introduced it into your target cells with Cas9. Hooray! Now it’s time to make sure your genome edits went according to plan. In this blog post we’ll explain how to verify that your cells were appropriately edited for your desired mutation - insertion, deletion, or site-directed knock-in. We’ll also discuss what to do if your editing efficiency isn’t as great as you would like, you have options!

What to know in advance
Before validating your edit, it’s important to know a few things about your target cells and the desired edits. First, you will need to know what the ploidy of your target site is. If you are trying to introduce a homozygous mutation, it’s helpful to know how many copies you are looking for. Secondly, categorize your edit – is it any frameshift mutation? A specific insertion or deletion? A knock-in? A base edit? Once you know exactly what you are looking for and how many of that thing you are looking for, you are ready to begin the screening process.
Want to set yourself up for a screening success? Before you start analyzing individual clones, it’s wise to quickly assess whether a significant number of cells have been edited. For many experiments, the end goal is to generate a clonal cell line, so it’s important to verify that you won’t need to screen hundreds of clones looking for your edit of interest. For all of the screening strategies outlined below, the technique can be used as a preliminary validation on your bulk population. This way you can ensure you aren’t looking for a needle in a haystack before going on to generate and screen individual clones. If a clonal line isn’t your end goal, then a single, bulk validation may be enough to proceed with your experimental plan.
Validating knockout mutations
When a knockout cell line is desired, any mutation that introduces a frameshift in the coding region of interest will get the job done. In other words, any insertion or deletion that isn’t a multiple of three will do. A popular screening method for knockouts is Tracking of Indels by Decomposition (TIDE) (Brinkman et al., NAR). This technique uses the trace file generated in Sanger sequencing and a simple algorithm to quantify editing efficiency by insertion and deletion frequency.
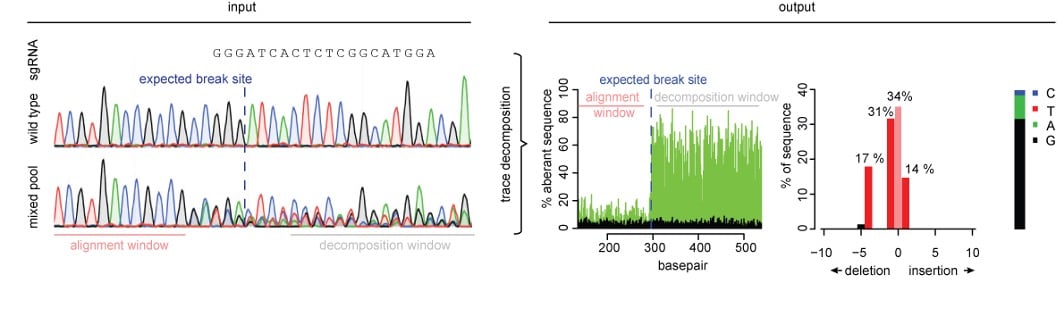 |
| Fig. 1: Overview of TIDE. The input is sequencing trace files of WT and edited cell lines. Then decomposition analysis is performed yielding a graph of edits in the input population or clone. |
How to perform a TIDE
First, use PCR to amplify the target region of interest. Then Sanger sequence the PCR product of both an unedited population and your Cas9-targeted cells. Simply upload both trace files to the TIDE tool online along with your sgRNA sequence. The TIDE software will generate a graph representing all insertions and deletions identified within a given genomic window, along with the estimated editing frequency of your sgRNA-Cas9 combo.
Pro tip: When designing your PCR, ensure there is at least ~200 base pairs of sequence flanking the editing site on either side of the amplicon.
Based on the frequency of out-of-frame edits, you can determine if it is worthwhile to isolate individual clones from that population. You can additionally calculate the minimum number of clones you would have to screen to identify a knockout clone. For example, if your cell line is diploid (2N) and your out of frame editing frequency is 50%, then you could infer that approximately 25% of cells are likely -/-, 50% are -/+, and 25% are +/+ for your gene of interest. Thus, to find a null clone, you should screen, at the very least, 4 clones.
Did you use a dual guide system to generate your knockout? Then you’re in luck! You don’t need to sequence your PCR to visualize an edit. Most dual guide systems generate a deletion large enough that it can be visualized by running your PCR on a gel. It’s still paramount that you sequence clones to verify that the deletions introduced are out of frame but finding clones with deletions is faster when using a dual guide system.
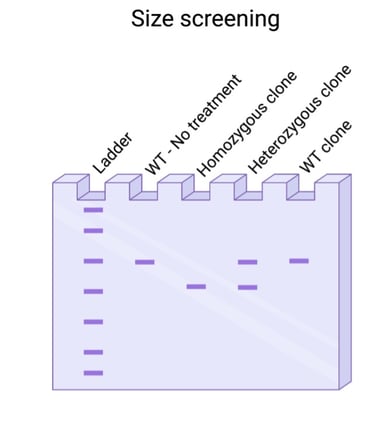 |
|
Fig. 2: Size screening for large deletion events. |
Validating knock-in mutations
You’ve introduced a donor template along with your gRNA and Cas9 combo and are hoping for homology-directed repair events, or maybe you’re performing a single base pair edit with a prime editor. What next? When generating a knock-in mutation, the best screening method depends on what your desired edit is.
Large knock-ins
Are you introducing a large insertion, something 20 base pairs or bigger? Then screening by PCR may be the way to go. Design a PCR to flank your desired edit site and run the PCR product on a gel to look for size shifts. Once you’ve identified homozygous clones, however, sequence confirmation is still needed to verify the correct sequence was inserted or deleted accordingly.
Pro tip: make sure your PCR amplicon isn’t too large so that you can easily visualize the size shift you anticipate. A good rule of thumb is to keep your product to knock-in size ratio below 10:1.
Small knock-ins
Is your knock-in edit small, with only a few base pairs modified? In these cases, it’s best to do some planning in the design phase of your experiment for a screening strategy. Restriction enzyme screening is sensitive to single base pair changes, but the edit must occur at an existing restriction enzyme recognition sequence or generate a new recognition sequence. In these cases, you can screen using PCR and a restriction enzyme digest.
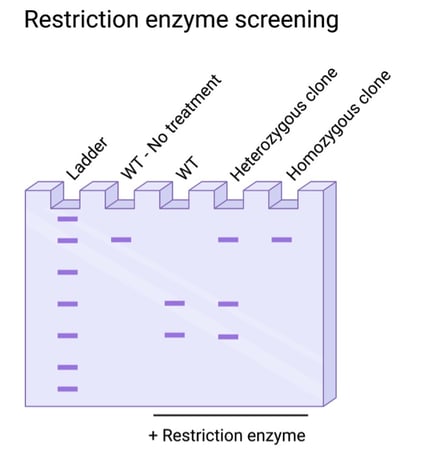 |
|
Fig. 3: Restriction enzyme screening for resistivity to digestion, indicating targeted edits. |
Interested in this method, but your edit doesn’t affect any endogenous restriction enzyme sites? Never fear! You can introduce a silent, passenger edit to serve as a proxy for your edit of interest. Using the same gRNA and donor, you can deliver a silent mutation in a neighboring nucleotide along with your desired edit. This silent mutation should be designed solely for restriction enzyme sensitivity or resistivity, and thus be a marker for edited cells. As with all other methods, you’ll need to further validate the specific mutations using sequencing confirmation.
All knock-ins
A method applicable to knock-ins both large and small is a software very similar to TIDE known as TIDER (Tracking of insertions, deletions, and recombination events). TIDER can screen for specific homology-directed repair events and small edits (Brinkman, et al., NAR). It works just like TIDE (outlined above), except that it uses a 3rd sequencing trace file from the donor DNA. This method can precisely estimate editing frequency in a bulk population and also identify edits in individual clones. The drawback is that you must generate a DNA molecule with homology to your genome edit to mimic what an edited sequence would look like (the 3rd sequencing trace). While it requires a little extra wet lab work, this is still a great option, especially if the options outlined above aren’t realistic.
Check Out Our Post on Sequencing Options for CRISPR Genotyping
NGS and detecting off target effects
While pricier, NGS can be used to quantitatively assess genome edits in your target sequence and other regions of the genome. The main advantage of NGS is that it allows you to simultaneously validate edits and look for off-target changes. This method requires a set of control cells to compare the sequencing reads from your edited sample to an untreated population. Software such as CRISPResso can help with the data analysis.
If NGS is not an option, you can still detect off-target effects with Sanger sequencing, provided you have some idea of where in the genome you might expect off target editing to occur. Luckily, in silico prediction analyses like CRISPRitz or CRISPOR can help narrow down the list of sites where off target editing may occur, allowing you to Sanger sequence a manageable number of the higher probability locations.
If you’re worried about off-target effects, you can consider using a version of Cas9 that has been specifically engineered for reduced off target editing, such as SpCas9-HF1, eSpCas9(1.1), or HypaCas9. Read more about Cas9 options in our blog post here.
Dealing with low editing frequency
Did your editing experiment not go to plan? Don’t despair, you have options! If you think delivery of Cas9 or gRNA is the issue, check out or blog on optimizing this step, or consider enriching for transfected cells by flow sorting or drug selection, assuming your reagents are amenable to this. If delivery isn’t an issue, you may need to select an alternative gRNA. Make sure to check out our validated gRNAs to avoid rolling the dice on a new gRNA in the future!
References and Resources
References
Eva K. Brinkman, Tao Chen, Mario Amendola, Bas van Steensel. Easy quantitative assessment of genome editing by sequence trace decomposition. (2014) Nucleic Acids Research Volume 42, Issue 22, Page e168. https://doi.org/10.1093/nar/gku936
Eva K. Brinkman, Arne N. Kousholt, Tim Harmsen, Christ Leemans, Tao Chen, Jos Jonkers, Bas van Steensel. Easy quantification of template-directed CRISPR-Cas9 editing. (2018) Nucleic Acids Research Volume 46, Issue 10, Page e58. https://doi.org/10.1093/nar/gky164
Cancellieri S, Canver MC, Bombieri N, Giugno R, Pinello L. CRISPRitz: rapid, high-throughput and variant-aware in silico off-target site identification for CRISPR genome editing. (2019) Bioinformatics 36:2001–2008. https://doi.org/10.1093/bioinformatics/btz867
Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud J-B, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly J-S, Concordet J-P. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. (2016) Genome Biol 17: https://doi.org/10.1186/s13059-016-1012-2
Find more CRISPR Resources at Addgene:
- Read our CRISPRGuide
- Get practical help from our CRISPR Protocols
- Find gRNA Design Tools
- Read other CRISPR blog posts
Find CRISPR Plasmids at Addgene

Topics: CRISPR, CRISPR 101





Leave a Comment