 Addgene is proud to announce that we recently acquired the ability to distribute plasmids with the piggyBac™ transposon. These plasmids, when combined with a source of piggyBac™ transposase (available from a licensed distributor) allow you to quickly transfer a DNA sequence from the transposon vector to one of many TTAA sequences distributed throughout the genome. We encourage you to deposit your piggyBac™ transposon vectors with us to help us expand this useful resource. While Addgene cannot distribute plasmids with the piggyBac™ transposase itself, please read on to learn more about this exciting technology from the folks at Transposagen.
Addgene is proud to announce that we recently acquired the ability to distribute plasmids with the piggyBac™ transposon. These plasmids, when combined with a source of piggyBac™ transposase (available from a licensed distributor) allow you to quickly transfer a DNA sequence from the transposon vector to one of many TTAA sequences distributed throughout the genome. We encourage you to deposit your piggyBac™ transposon vectors with us to help us expand this useful resource. While Addgene cannot distribute plasmids with the piggyBac™ transposase itself, please read on to learn more about this exciting technology from the folks at Transposagen.
This post was contributed by Amar Singh and Mathew Goodwin from Transposagen
What is the piggyBac™ transposon?
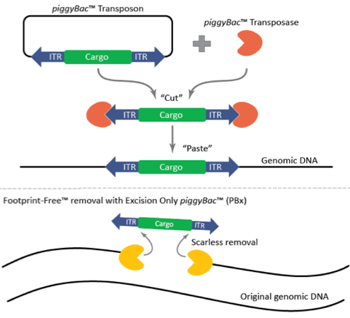
The piggyBac™ transposon was originally identified in moths in 1983, but wasn’t successfully used for genetic manipulation in mammalian cells until 2005 [1]. Like other transposons, piggyBac™ has two components, a transposon and a transposase. The piggyBac™ transposase facilitates the integration of the transposon specifically at ‘TTAA’ sites randomly dispersed in the genome. The predicted frequency of ‘TTAA’ in the genome is 1 in every 256 base-pairs of DNA sequence, making it very useful for genetic engineering approaches. Most importantly for this technology, however, is that the transposase also enables the excision of the transposon in a completely seamless manner, leaving no sequences or mutations behind. Furthermore, piggyBac™ offers a large cargo-carrying capacity (over 200 kb has been demonstrated) with no known upper limit [1]. The piggyBac™ technology can be used for numerous applications, including transgenesis, gene-trap screens, and gene editing.
How does the piggyBac™ system make it easier to generate transgenic cell lines?
One of the major uses of the piggyBac™ system is to create stable transgenic cell lines. The traditional method of making stable cell lines is to transfect, select, and isolate clones. This procedure is cumbersome and inefficient, as it relies on random integration efficiencies that may be low in the cell line of interest. The piggyBac™ system allows for significantly enhanced random integration efficiencies that are accomplished with transfections and are therefore virus-free. This is achieved using the piggyBac™ transposase, which facilitates direct integration of the donor-transposon (carrying your cargo of interest) into random ‘TTAA’ sites throughout the genome. When using piggyBac™, the cargo is inserted into the genome fully intact, while, with a standard DNA plasmid transfection, a double-stranded break may occur in your cargo. Additionally, with piggyBac™, the number of integration copies can be tightly controlled by titrating the transposase to transposon ratios. High ratios of transposase to transposon result in a greater number of integrations per cell and a super-piggyBacTM can be used to get an even greater number of integrations. It should be noted, however, that piggyBac™ integration efficiency is likely to be cell line-dependent, and should be tested in the cell line of interest. Importantly, by varying these ratios, you can either maximize or minimize expression levels of your gene of interest, as needed. Controlling the expression levels of your gene may be particularly useful when the gene being expressed has threshold effects, such as with signaling molecules or transcription factors, or when the gene is constitutively-active or a dominant-negative mutant. Reducing the copy number can also mitigate the deleterious effects of expression of certain genes, such as those that have negative impacts on fitness.
One consideration when making a transgenic cell line with the piggyBac™ system is that it has a preference for integration at regions of open chromatin, especially at promoter or exonic regions. If the cell’s chromatin structure undergoes significant rearrangement (such as when a stem cell initiates differentiation), transgene silencing may occur (as it would with any integrated transgene). To overcome this obstacle, a positive selection cassette can be incorporated, and used during the differentiation process. Moreover, a splinkerette-PCR assay may be used to map the specific genetic loci of integration [8].
 |
|
Figure 1: Mechanism of the piggyBac™ transposase/transposon gene modification system and the scarless, Footprint-Free™ removal of cargo with the excision-only piggyBac™ transposase. Image courtesy of Transposagen. |
How does piggyBac™ enhance gene editing?
Gene editing can now be accomplished in a more cost-effective and efficient manner than ever before, thanks to the development of site-specific nucleases (CRISPR/Cas9, TALENs, and ZFNs). However, several challenges still remain, such as making gene edits completely seamless (i.e., so no other genetic alterations have occurred) and isolating the specific cells that have the edit of interest. Fortunately, by combing nuclease technology with the piggyBac™ system, these challenges can be considerably reduced.
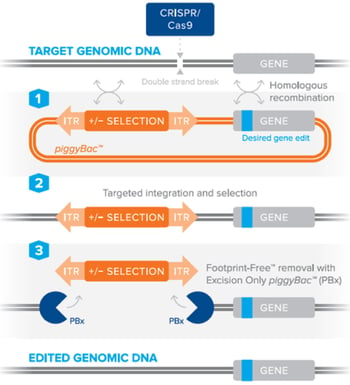
The piggyBac™ transposon system can be combined with CRISPR/Cas9 to efficiently perform genetic manipulations in animal models or cells lines, including stem cells, without leaving behind any accessory DNA sequences [2], [3]. Simply put, the piggyBac™ transposon containing a selection marker is included in a standard homology directed repair template to facilitate the selection of cells containing your desired edit. The selection is marker is then seamlessly removed using the piggyBac™ transposase. This can be best achieved by utilizing a piggyBac™ transposon in a donor plasmid carrying the DNA edit of interest, along with a site-specific nuclease (such as CRISPR/Cas9) to create a double-stranded DNA break. After DNA cleavage, the piggyBac™ donor is used by the cell’s host machinery for DNA repair by homologous recombination, which results in the incorporation of your specific edit into the host genome. Through the use of an excision-only piggyBac™ transposase (PBx), which contains mutations making it competent for excision, but defective for integration [4], the donor cassette is removed leaving behind no accessory transposon sequences. The use of PBx is particularly important to avoid any possible random re-integration from the piggyBac™ donor, which is typically found with the wild-type piggyBac™ transposase enzyme [4]. By using a donor plasmid containing a negative selection marker such as thymidine kinase, a researcher may subsequently kill off any cells that still contain the donor plasmid after excision by PBx, thereby leaving behind only those cells with a Footprint-Free™ genome edit.
 |
|
Figure 2: Editing DNA using the piggyBac™ transposon selection technology using a site-specific nuclease to stimulate homologous recombination and Footprint-Free™ removal of the selection cassette by excision-only piggyBac™. Image courtesy of Transposagen. |
How does the piggyBac™ gene editing system compare with other methods of gene editing?
There are primarily two other ways to create specific point mutations by gene editing. First, instead of using piggyBac™, you could use recombinase technology (Cre/loxP) to remove the donor cassette. While this method can be effective, a loxP site is left behind in the genome following donor removal, which alters the endogenous DNA sequence and has been suggested to cause cytotoxicity [5]. Second, instead of using a donor-plasmid, you could use a single-stranded oligodeoxynucleotide (ssODN) as the donor [6]. The problem with this approach is that, since there is no selection, you will need to screen somewhere between one hundred and one thousand clones to find a cell line with the edit of interest. With the larger piggyBac™ donor vector, it is possible to make the ITR and selection cassette disrupt the target site for the nuclease thereby preventing re-cutting. Transposase removal of the cassette later restores the target sequence. This allows researchers to avoid making unnecessary base pair changes as would be the case with ssODNs.
One minor drawback with using PBx for excision is its efficiency. While the efficiency of excision with PBx has been shown to be higher than that of the wildtype piggyBac™ transposase or hyperactive transposase mutants [7], it is generally not as efficient as Cre recombinase. However, the use of negative selection largely overcomes this obstacle. Furthermore, we have found that transfecting with PBx mRNA can boost efficiency over transfecting with plasmids in some cell lines, most likely due to DNA toxicity. More importantly, unlike Cre recombinase, the PBx is highly specific, and has not been shown to cause genomic abnormalities when expressed. This lack of off-target effects is likely due to the fact that PBx relies on the inverted terminal repeats (ITRs) found on the piggyBac™ donor for its activity.
In summary, the piggyBac™ transposon is a powerful technology that may be used for a variety of research applications, including making transgenic cell lines and genetically-modified cell lines. Furthermore, the use of piggyBac™ for gene editing allows you to efficiently perform seamless genetic manipulations, while avoiding the pitfalls common to other approaches.

Many thanks to our Guest Bloggers Amar Singh and Mathew Goodwin from Transposagen!
 Dr. Amar Singh is a Senior Scientist at Transposagen. He has a long-standing interest in molecular and cellular biology and brings 10 years of research experience with stem cells to Transposagen's cell engineering team.
Dr. Amar Singh is a Senior Scientist at Transposagen. He has a long-standing interest in molecular and cellular biology and brings 10 years of research experience with stem cells to Transposagen's cell engineering team.
 Mathew Goodwin is a Technical Sales Specialist at Transposagen. As a gene editing enthusiast, he has spent over 6 years in the lab developing and using a variety of gene editing technologies including Zinc Fingers, TALENs, and CRISPR-Cas9 nucleases. Mathew applies his specialized knowledge to help researchers identify the right technologies and strategies for their projects.
Mathew Goodwin is a Technical Sales Specialist at Transposagen. As a gene editing enthusiast, he has spent over 6 years in the lab developing and using a variety of gene editing technologies including Zinc Fingers, TALENs, and CRISPR-Cas9 nucleases. Mathew applies his specialized knowledge to help researchers identify the right technologies and strategies for their projects.
References
1. L. E. Woodard and M. H. Wilson, “piggyBac-ing models and new therapeutic strategies.,” Trends Biotechnol., vol. 33, no. 9, pp. 525–33, Sep. 2015. PubMed PMID: 26211958. PubMed Central PMCID: PMC4663986.
2. A. M. Singh, V. V Adjan Steffey, T. Yeshi, and D. W. Allison, “Gene Editing in Human Pluripotent Stem Cells: Choosing the Correct Path.,” J. stem cell Regen. Biol., vol. 1, no. 1, Jan. PubMed PMID: 26702451. PubMed Central PMCID: PMC4686154.
3. A. M. Singh, D. W. Perry, V. V. A. Steffey, K. Miller, and D. W. Allison, “Decoding the Epigenetic Heterogeneity of Human Pluripotent Stem Cells with Seamless Gene Editing.,” Methods Mol. Biol., Apr. 2016. PubMed PMID: 27075976.
4. X. Li, E. R. Burnight, A. L. Cooney, N. Malani, T. Brady, J. D. Sander, J. Staber, S. J. Wheelan, J. K. Joung, P. B. McCray, F. D. Bushman, P. L. Sinn, and N. L. Craig, “piggyBac transposase tools for genome engineering.,” Proc. Natl. Acad. Sci. U. S. A., vol. 110, no. 25, pp. E2279–87, Jun. 2013. PubMed PMID: 23723351. PubMed Central PMCID: PMC3690869.
5. D. Grégoire and M. Kmita, “Recombination between inverted loxP sites is cytotoxic for proliferating cells and provides a simple tool for conditional cell ablation.,” Proc. Natl. Acad. Sci. U. S. A., vol. 105, no. 38, pp. 14492–6, Sep. 2008. PubMed PMID: 18787116. PubMed Central PMCID: PMC2567143.
6. S. Radecke, F. Radecke, T. Cathomen, and K. Schwarz, “Zinc-finger nuclease-induced gene repair with oligodeoxynucleotides: wanted and unwanted target locus modifications.,” Mol. Ther., vol. 18, no. 4, pp. 743–53, Apr. 2010. PubMed PMID: 20068556. PubMed Central PMCID: PMC2862519.
7. A. L. Firth, T. Menon, G. S. Parker, S. J. Qualls, B. M. Lewis, E. Ke, C. T. Dargitz, R. Wright, A. Khanna, F. H. Gage, and I. M. Verma, “Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs.,” Cell Rep., vol. 12, no. 9, pp. 1385–90, Aug. 2015. PubMed PMID: 26299960. PubMed Central PMCID: PMC4559351.
8. A. G. Uren, H. Mikkers, J. Kool, L. van der Weyden, A. H. Lund, C. H. Wilson, R. Rance, J. Jonkers, M. van Lohuizen, A. Berns, and D. J. Adams, “A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites.,” Nat. Protoc., vol. 4, no. 5, pp. 789–98, Jan. 2009. PubMed PMID: 19528954. PubMed Central PMCID: PMC3627465.
Additional Resources on the Addgene Blog
- Learn about the Sleeping Beauty Transposase
- Read up on Genome Engineering with FLEx Vectors
- Learn how Homology Directed Repair Is Used for Genome Editing
Resources on Addgene.org
- Find Vectors with the piggyBac™ Transposon
- Catch up on Everything CRISPR with Our CRISPR Guide
- Deposit Your piggyBac™ Vectors!
Topics: CRISPR, Other CRISPR Tools





Leave a Comment