This post was contributed by Jae Lee and Pantelis Tsoulfas of the Department of Neurological Surgery at the University of Miami.
The beginning of this century has seen some major advances in light microscopy, particularly related to the neurosciences. These developments in microscopy coupled with techniques that make tissues transparent are enabling microscopes to visualize the cellular architecture of whole tissues in 3D with unprecedented detail. One of these advances in microscopy has been light sheet fluorescence microscopy (LSFM). The underlying method was developed in 1902 by Richard Zsigmondy and Henry Siedentopf to enhance the microscopic resolution for studying colloidal gold (1). The method was based on using a thin plane (sheet) of light generated by sunlight to observe single gold particles with diameters less than 4nm.
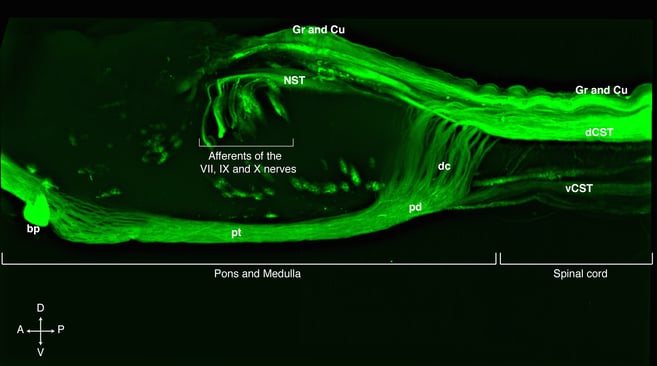 Lateral view of a cleared rat pons, medulla and the first segments of the spinal cord showing the cortico spinal tract, CST, spinal cord and cranial nerve afferents labelled with a AAV8 expressing eGFP under the human ubiquitin C promoter. Anatomical abbreviations: Gr= Gracile tract, Cu= Cuneate tract, dCST= dorsal cortico spinal tract, vCST= ventral cortico spinal tract, dc= decussation of the CST, pd= pyramid, pt= pyramidal tract or CST traversing pons and medulla, bp= basilar pons, NST nucleus of the solitary tract. The arrows point the direction of the specimen, A= anterior, P= posterior, D= dorsal, V= ventral
Lateral view of a cleared rat pons, medulla and the first segments of the spinal cord showing the cortico spinal tract, CST, spinal cord and cranial nerve afferents labelled with a AAV8 expressing eGFP under the human ubiquitin C promoter. Anatomical abbreviations: Gr= Gracile tract, Cu= Cuneate tract, dCST= dorsal cortico spinal tract, vCST= ventral cortico spinal tract, dc= decussation of the CST, pd= pyramid, pt= pyramidal tract or CST traversing pons and medulla, bp= basilar pons, NST nucleus of the solitary tract. The arrows point the direction of the specimen, A= anterior, P= posterior, D= dorsal, V= ventral
Developments in light sheet fluorescence microscopy
Modern light sheet fluorescence microscopy was first pioneered by Voie and colleagues and originally named orthogonal-plane fluorescence optical sectioning (OPFOS) (2). Arne Voie, David Burns and Francis Spelman focused a laser beam into a thin sheet to illuminate a fluorescent sample and captured the reflected light using a different objective lens oriented perpendicular to the plane of illumination (i.e. light sheet). This is in contrast to confocal laser microscopy where the laser and the reflected light travel through the same objective lens. Using this method, these authors were able to reconstruct a cleared guinea pig cochlea in 3D. The basic configuration of the instrument used in this reconstruction lay the foundation for all subsequent versions of LSFM microscopes (3,4).
After this initial application, Stelzer’s group described the single-plane or selective-plane illumination microscope (SPIM). In order to improve axial resolution and reduce phototoxicity for imaging live specimens, they combined light sheet microscopy with sequential multiple-view reconstruction (5). In order to image large tissue such as cleared rodent brain, the Ultramicroscope was developed using a stereomicroscope to capture large field-of-views (6). For more comprehensive reviews on several other LSFM configurations’ various features see (4,7).
Light sheet fluorescence microscopy vs. confocal microscopy
In confocal microscopes, optical sectioning of a specimen is based on discriminating the out-of-focus reflected light by using a pinhole. However, the excitation light excites all of the fluorophores as it passes through the specimen, which often leads to photobleaching. Furthermore, point-by-point scanning along the entire specimen makes imaging too slow and unwieldy for large specimens that are millimeters in thickness (8,9). In addition, there is a drop in image brightness with increasing depth caused by light scattering and absorption. Finally, the photomultipliers used to detect light in confocal microscopes are less efficient than the modern cameras used in LSFM.
In LSFM the laser light sheet, typically 2-6 microns, illuminates only one thin plane of the sample surrounding the focal plane of the detection lens and thus there is no out of focus light. Therefore, there is much less photo-bleaching or photo-damage than in conventional laser scanning microscopy . Since image acquisition is performed using cameras with electron multiplying couple-charged device (EMCCD) or complementary metal-oxide semiconductor (CMOS) sensors, it takes only a few milliseconds to acquire one image, thereby significantly reducing the time it takes to image through a large z-stack (8). One limitation of LSFMs using single side illumination, especially with large tissue, is that any obstacles (e.g. air bubbles or high concentration of fluorophores) cast shadows along the illumination path. The shadows appear as stripes in the acquired image (5). A way to get around this problem is to illuminate and acquire images of a sample from two opposing sides and merge the images, such as in the Ultramicroscope. In addition, adjusting the thickness of the laser sheet and using multi-view imaging can provide enhanced resolution (8,9). Thus, LSFM is ideal for 3D reconstruction of clear specimens and for in vivo imaging of transparent organisms.
Advances in tissue clearing
Similar to light microscopy, there have been significant advances in tissue clearing techniques during this century. The first successful attempt to render fixed anatomical preparations transparent was done by Walter Spalteholtz (10). He used a solution of benzyl alcohol and methyl salicylate which was later modified by others to produce Murray’s clear solution. Murray's clear (11) was mostly used to study the development of vertebrate embryos. The first 3D reconstructions of clear embryos were obtained by using optical projection tomography (OPT) (12) and later Hans-Ulrich Dodt’s lab pioneered the use of light sheet microscopy on cleared whole-mount specimens, such as whole mouse brains expressing green fluorescent protein (GFP) (6).
The Dodt and Frank Bradke laboratories later developed the 3DISCO (3-dimensional imaging of solvent cleared organs) method of imaging rodent brains and spinal cords by combining Ultramicroscopy with tetrahydrofuran-based tissue clearing (13,14). A few years later, Karl Deisseroth’s lab developed CLARITY, which is a hydrogel-based method that also allows antibody penetration for immunohistochemical labeling in whole tissue (15). CLARITY is also compatible with LSFM, a method termed COLM (clarity-optimized light-sheet microscopy) (16). Both 3DISCO and CLARITY have relied on the use of transgenic mice in which a subpopulation of neurons are brightly labeled with GFP (e.g. Thy1-YFP-H mice). However, we recently developed AAV (adeno-associated virus)-based fluorescent labeling methods that can be used with 3DISCO to image non-transgenic animals such as rats (17). In addition, the use of viruses allows much better spatiotemporal control over labeling methods as compared to transgenic animals (17). These viral plasmids are available through Addgene. The combination of LSFM with multiple tissue clearing strategies and neuronal labeling methods will greatly aid our understanding of the structure-function relationship of the central nervous system under normal and diseased conditions.
Thank you to our guest bloggers, Jae Lee and Pantelis Tsoulfas.
Jae K. Lee is an assistant Professor at the University of Miami Miller School of Medicine. Find plasmids from the Lee Lab here.
Pantelis Tsoulfas is an Associate Professor at the University of Miami Miller School of Medicine. Find plasmids from the Tsoulfas lab here.
References:
- H. Siedentopf, and R,. Zsigmondy. (1902) Ann. Phys. 315, 1 – 39.
- A. H. Voie, D. H. Burns, F. A. Spelman. (1993) J. Microsc. 170, 229 – 236. Pubmed.
- P.A Santi. (2011). Journal of Histochemistry & Cytochemistry, 59, 129-138. Pubmed.
- J. Huisken, and D.Y.R. Stainier. (2009). Development, 136, 1963–1975. Pubmed.
- J. Huisken, J. Swoger, F. Del Bene, J. Wittbrodt, Stelzer E.H.K. (2004) Science, 305, 1007-1009. Pubmed.
- H.-U. Dodt, U. Leischner, A. Schierloh, N. Jährling, C.P. Mauch, K. Deininger, et al. (2007). Nature Methods, 4, 331–336. Pubmed.
- P. Keller, M.B. Ahrens. (2015) Neuron, 85, 462-483. Pubmed.
- E.H.K. Stelzer. (2015). Nature Methods, 12, 23–26. Pubmed.
- M. Weber, M. Mickoleit, J. Huisken. (2014) Methods Cell Biol.123, 193-215. Pubmed.
- W. Spalteholz. (1914) Uber das Durchsichtigmachen von menschlichen und tierischen Praparaten. Leipzig: S. Hierzel.
- J.A Dent, A.G. Polson, M.W. Klymkowsky. (1989) Development, 105.61-74. Pubmed.
- J. Sharpe, U. Ahlgren, P. Perry, Hill B, Ross A, J. Hecksher-Sorensen, R. Baldock, D. Davidson. (2002) Science, 296, 541-545. Pubmed.
- A. Ertürk, C.P. Mauch, F. Hellal, F. Förstner, T. Keck, K. Becker, et al. (2011). Nature Medicine, 18, 166–171. Pubmed.
- A. Ertürk, K. Becker, N. Jährlin, C.P. Mauch, C.D. Hojer, J.G. Egen, F. Hellal, F. Bradke, M. Sheng, H.-U. Dodt. (2012). Nature Protocols, 7(11), 1983–1995. Pubmed.
- K. Chung, J. Wallace, S.Y. Kim, S. Kalyanasundaram, A.S. Andalman, T.J. Davidson et al. (2013) Nature, 497, 332-337. Pubmed.
- R. Tomer, L. Ye, B. Hsueh, K. Deisseroth. (2014) Nat Protoc., 9, 1682-1697. Pubmed.
- C. Soderblom, D.-H. Lee, A. Dawood, M. Carballosa, et al, (2015) eNeuro, March/April 2015, 2(2) e0001-15.2015. Pubmed.

Topics: Fluorescent Proteins, Fluorescent Imaging





Leave a Comment