Originally published Nov 30, 2017 and updated Jul 31, 2020.
Cas13 enzymes are quickly becoming major players in the CRISPR field. Just a year after Feng Zhang’s lab identified Cas13a (C2c2) (Abudayyeh et al., 2016) as a RNA-targeting CRISPR enzyme, they adapted Cas13b for precise RNA editing (Cox et al., 2017). This new system, termed REPAIR (RNA editing for programmable A to I (G) replacement) is the first CRISPR tool for RNA editing. Two years after that, the lab published a paper on an RNA editor that allows C to U edits (RESCUE). We’ll walk through how these tools were developed and potential ways you can use it in your research.
RNA editing advantages
RNA editing has multiple advantages over more traditional DNA editing systems; first, RNA editing doesn't require homology-directed repair (HDR) machinery, and could thus be used in non-dividing cells. Cas13 enzymes also don’t require a PAM sequence at the target locus, making them more flexible than Cas9/Cpf1. Some Cas13 enzymes prefer targets with a given single base protospacer flanking site (PFS) sequence, but orthologs like LwaCas13a do not require a specific PFS. Cas13 enzymes do not contain the RuvC and HNH domains responsible for DNA cleavage, so they cannot directly edit the genome. A Cas13-based RNA editing system would likely be reversible and would avoid genomic off-targets or indels introduced through non-homologous end joining (NHEJ).
Designing an RNA editor
The Zhang lab envisioned a two-component RNA editor: a Cas13 enzyme fused to an RNA adenosine deaminase (ADAR). Such a system would convert adenine to inosine, which the translational machinery treats like guanine. This RNA editor would permit point mutations in RNA which could recapitulate or rescue known pathogenic alleles, or introduce a premature stop codon to render an RNA nonfunctional.
In their quest to build a robust RNA editor, the lab started with the Cas13 scaffold, testing a whopping 21 Cas13a orthologs, 15 Cas13b orthologs, and 7 Cas13c orthologs. They hoped to find a stably folded ortholog that cleaved RNA robustly, unlike LwaCas13a, which must be stabilized by monomeric superfolded GFP and averages only ~50% RNA knockdown. In initial tests, Cas13b from Prevotella sep. P5-125 (PspCas13b) yielded 62.9% average knockdown, and they chose this enzyme for further studies. PspCas13b does not require a PFS, and it is sensitive to mismatches in target RNA from bases 12-26 of a 30 nt target sequence.
For the editing portion of the protein, they examined ADAR1 and ADAR2, which deaminate adenosine to inosine in RNA, creating a functional A->G change. They fused ADAR deaminase domains (ADARDD) to dPspCas13b, but observed low RNA editing. To increase A->G editing, they employed hyperactive ADAR constructs, like ADAR2DD(E488Q). They also adjusted the structure of the guide RNA by introducing a C opposite the target A to be edited. This change specifies the edit to be made when multiple As are present in the gRNA spacer, as ADAR will preferentially edit an adenine if the template has a cytosine mismatch at that position.
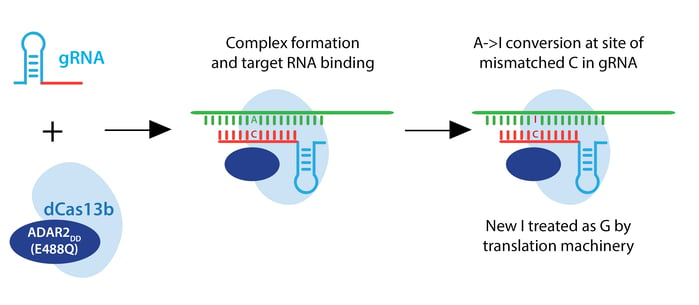
PspCas13b-ADAR2DD(E488Q) displayed robust editing with various spacer lengths from 30-84 nucleotides, and designated this system REPAIRv1. Using next-generation sequencing, they confirmed A->I editing and found that 50 nt spacers increase editing efficiency but also increase off-target editing within the target window, possibly due to longer stretches of duplexed RNA. Using an ADAR2DD catalytic mutant, they showed that editing is mediated by ADAR2DD, not PspCas13b.
Find the plasmids for the RESCUE RNA editors here!
Testing and improving RNA editing from the Cas13b REPAIR system
To test the robustness of REPAIRv1, the lab targeted 34 pathogenic G->A mutations from the ClinVar database. They successfully edited 33/34 sites with a maximum of 28% editing efficiency in HEK293T cells, as assessed using RNA-seq. Since the REPAIRv1 machinery is too large to fit into adeno-associated viral vectors, they tested truncated ADARDDs to see if they could shrink the construct. They identified ADAR2DD(delta984-1090), which decreases the REPAIRv1 construct size from 4,473 bp to 4,152 bp without decreasing editing efficiency.
Although REPAIRv1 knockdown is more precise than shRNA knockdown, it still displays substantial off-target activity. By mutating residues in ADAR2 that interact with duplex RNA, they hoped to destabilize ADAR-RNA binding to decrease off-targets. Of the tested constructs, ADAR2DD(E488Q/T375G) (REPAIRv2) had the highest on-target efficiency and the lowest amount of off-target editing. In one direct transcriptome-wide comparison, REPAIRv2 reduced off-target sites from the 18,385 observed with REPAIRv1 to merely 20.
Expanding RNA Editing Capabilities through RESCUE
Two years later, the Zhang lab expanded the RNA editing toolkit to allow C to U edits using their RESCUE (RNA Editing for Specific C-to-U Exchange) system (Abudayyeh et al., 2019). To avoid some of the inherent drawbacks for RNA editing of natural cytidine deaminases, they evolved the adenine deaminase ADAR2DD to deaminate cytidine and used dRanCas13b (catalytically inactive Cas13) to target the cytosine deaminase. Three rounds of rational mutagenesis on the ADAR2DD residues that contact the RNA substrate resulted in 15% C-to-U editing activity. They then performed sixteen rounds of directed evolution across the full ADAR2DD to further optimize cytidine deaminase activity. While RESCUE can perform C-to-U edits, the authors note that it retains its adenine deaminase ability.
The authors then tested the RESCUE system’s ability to edit endogenous transcripts finding up to 42% editing efficiency. They note that RESCUE is most active in a 30 nt guide when C or U base-flips are present across from the target base. However, they saw that in addition to the targeted C-to-U edit, RESCUE had A-to-I off-target edits around the targeted nucleotide. Introduction of guanine-mismatches in the guide across from these off-target edits helped to minimize local off-target editing by RESCUE. To address both C-to-U and A-to-I transcriptome-wide off target effects observed in whole-transcriptome RNA-sequencing, the authors performed rational mutagenesis and identified that a S375A mutation in RESCUE (named RESCUE-S) offered ~76% on-target C-to-U editing while reducing off-target C-to-U edits by ~45% and off-target A-to-I edies by ~94% compared to RESCUE.
Addressing the potential use of RESCUE in therapeutic applications, the authors tested various dRanCas13b truncations that would allow packaging the construct for viral delivery. They found C-terminal truncations allowed the same or improved editing ability in a RESCUE system small enough for viral delivery.
Find the RESCUE RNA editing plasmids here!
Future directions
Building on previous RNA targeting work with Cas13, REPAIR is the first CRISPR-based system to enable precise RNA editing. The addition of RESCUE further expands the RNA editing toolkit and sequences that can be edited. As discussed above, the ability to edit RNA has multiple advantages, including reversibility and use in non-dividing cells. With the expanded targeting ability using both systems, the authors look forward to the expanded use of RNA editing in research and its potential therapeutic uses
We are also curious if REPAIR and RESCUE can function in a multiplexed manner to edit multiple RNAs. It’s clear that CRISPR RNA editors will open new doors for scientific research - how will you use these systems in your research?
Cary Valley contributed to updating this article.
References
Cox, David B.T., et al. “RNA editing with CRISPR-Cas13.” Science (2017):pii: eaaq0180. PMID: 29070703.
- Find plasmids from this paper at Addgene.
Abudayyeh, Omar O., et al. “RNA targeting with CRISPR-Cas13.” Nature 550(7675) (2017):280-284. PMID: 28976959.
- Find plasmids from this paper at Addgene.
Abudayyeh, Omar O., et al. "C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector." Science 353(6299) (2016):aaf5573. PubMed PMID: 27256883. PubMed Central PMCID: PMC5127784.
- Find plasmids from this paper at Addgene.
East-Seletsky, Alexandra, et al. "Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection." Nature 538(7624) (2016):270-273. PubMed PMID: 27669025.
- Find plasmids from this paper at Addgene.
Gootenberg, Jonathan S., et al. "Nucleic acid detection with CRISPR-Cas13a/C2c2." Science (2017): eaam9321. PubMed PMID: 28408723. PubMed Central PMCID: PMC5526198.
- Find plasmids from this paper at Addgene.
East-Seletsky, Alexandra, et al. "RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes." Mol Cell. 66(3) (2017):373-83. PubMed PMID: 28475872.
- Find plasmids from this paper at Addgene.
Abudayyeh, Gootenberg, et al. “A cytosine deaminase for programmable single-base RNA editing.” Science 2019 Jul 26;365(6451):382-386. PubMed PMID: 31296651
- Find plasmids from this paper at Addgene.
Additional Resources on the Addgene Blog
- Check out our CRISPR Featured Topic Page
- Learn about RNA Targeting with Cas13a
- Learn about CRISPR DNA Base Editing
Resources on Addgene.org
- Browse All CRISPR Plasmids
- Check out our CRISPR Guide Pages

Topics: CRISPR, CRISPR 101, Cas Proteins





Leave a Comment