This post was contributed by guest bloggers Dominik Paquet and Dylan Kwart from Ludwig-Maximilians-University in Munich and Marc Tessier-Lavigne’s lab at the Rockefeller University in NYC.
The CRISPR/Cas9 system is a versatile tool for precise gene editing in many organisms and model systems. We have used CRISPR/Cas9 extensively for the purpose of making sequence-specific changes in human induced pluripotent stem cells (iPSCs). The CRISPR/Cas9 complex is very efficient at introducing double stranded breaks (DSBs) into genomic DNA in many cell types and often results in biallelic modifications. Most commonly, DSBs are repaired by the nonhomologous end-joining (NHEJ) pathway, leading to nonspecific nucleotide insertions, deletions or other mutations, referred to as ‘indels’. While this is convenient for generating gene knockouts, NHEJ repair does not allow introduction of specific sequence changes.

To generate a specific sequence change the cell must undergo homology-directed repair (HDR), a distinct cellular DNA repair pathway. Accomplishing this usually involves simultaneously introducing a homologous DNA repair template, such as a single-stranded oligodeoxynucleotide (ssODN), which contains the intended sequence changes to be incorporated into the edited genome. In mammalian cells such as stem cells, HDR is relatively rare and DSBs are predominantly repaired by NHEJ. While other groups have focused on strategies to improve overall HDR rates, we have shown recently that desired genome-editing events by HDR can be generated more efficiently by (1) improving editing accuracy by preventing re-editing and (2) optimizing “cut-to-mutation distance”. Furthermore (3), we have developed a framework that, by combining these two strategies allows you to specifically incorporate homo- or heterozygous mutations.
1. Increasing HDR accuracy with CRISPR/Cas-blocking mutations
After genome editing with CRISPR/Cas9 in human cells was described in 2013 (Cong et al, Mai et al) we were very excited to set up a CRISPR-based editing platform in our lab. Our goal was to use the system to introduce disease-causing mutations in iPSCs (or correct them back to wild type), differentiate them into somatic cells affected by the disease and study what’s wrong with the cells. However, our excitement turned into frustration when we sequenced our cells and found how inefficiently our edits were being introduced. Although we were able to introduce the desired mutations in genes associated with dementia, such as APP and PSEN1, in most cases the intended HDR events were corrupted by unwanted indels on the same allele, probably due to concomitant NHEJ repair. At the PSEN1 locus, over 90% of the HDR edited alleles had unwanted insertions/deletions, therefore making those alleles useless for our studies. If one now also takes into account that HDR is rare, usually 2-5% of the edited cells in our hands, these combined pitfalls made it virtually impossible to find the 0.2-0.5% correctly edited alleles, when screening reasonable amounts of single cell clones. Furthermore, CRISPR/Cas9 editing is often bi-allelic, meaning that even if one allele is accurately edited, the other likely contains unwanted indels.
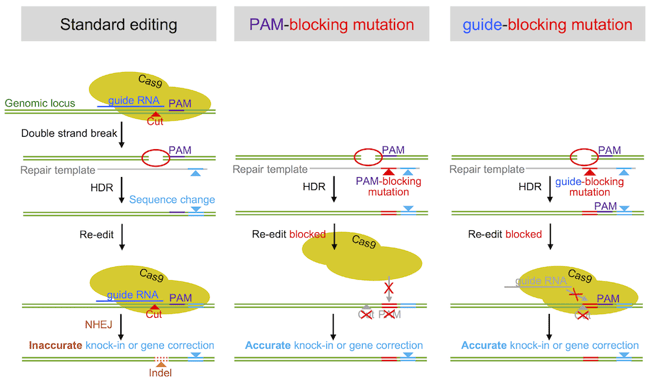
Following this observation, we tested a strategy that was discussed in the field at the time but had never been systematically analyzed: introducing silent CRISPR/Cas9 blocking mutations in either the PAM (changing one of the G’s to an A,T or C – but avoid NAG) or guide RNA target (“seed”) sequence. The idea was that these “blocking mutations” mutations prevent CRISPR from re-cutting the target sequence once the desired edit has been introduced.
We found that blocking mutations increased the accuracy of editing by up to 10-fold per allele. This is a 100-fold increase in editing accuracy if both alleles in a single cell are edited (Paquet, Kwart, et al 2016). For those of you involved in the tedious business of iPSC clone picking, a 100-fold decrease is the difference between picking 100 versus 10,000 clones! We would therefore strongly encourage any researcher to incorporate CRISPR/Cas-blocking mutations at both targeted alleles when gene editing.
 |
| Figure 1: Techniques to improve editing efficiency. |
But what’s better: PAM-blocking or guide RNA blocking mutations? This depends entirely on the locus you are editing, and if the blocking mutation needs to be silent. In our view, it’s always best to change the PAM site, however we have also demonstrated the efficiency of guide RNA blocking mutations. So if the locus does not allow blocking the PAM sequence (e.g. if it would lead to a missense or nonsense mutation), blocking the guide RNA binding site works well. However, since some guide RNAs may tolerate a mismatch in the binding sequence, we recommend changing several bases as close to the PAM as possible (see below to find out why). Please check our recent studies for details (Paquet, Kwart et al 2016, and Kwart, Paquet, et al 2017).
What if your locus does not allow you to use permanent blocking mutations at all, such as when you are editing a non-coding DNA region? To this end we developed a technology called CORRECT, which, by undergoing two rounds of gene-editing enables scarless introduction of an intended mutation while taking advantage of the improved efficiency provided by blocking mutations. Essentially, you:
Step 1 - Introduce the blocking mutation together with the intended sequence change and screen a few hundred clones to find the right one
Step 2 – Edit the cells a second time using a repair template that corrects the blocking mutation and again identify the right clone from a few hundred.
From these two steps, you’ll be screening a total of a few hundred clones - Still better than 10.000, right? (Kwart, Paquet, et al 2017).
Find Additional CRISPR Resources on Addgene's CRISPR Guide Page
2. Optimizing "cut-to-mutation distance"
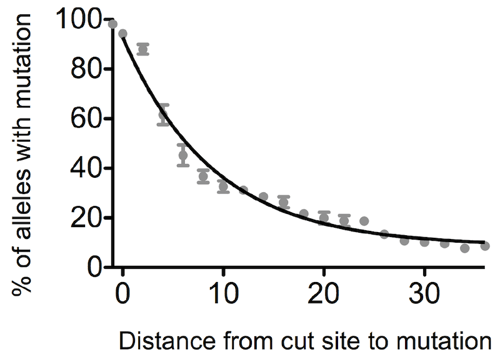
Even though we had controlled over-active Cas9 and repeated our experiments with blocking mutations, we still weren’t entirely happy with our editing outcomes and in many cases still didn’t get what we wanted. The Indels were gone from most HDR reads that we studied by deep sequencing, but interestingly (and to our frustration) in many cases we only saw incorporation of the blocking mutation. Despite the fact that both the blocking mutation and our intended mutation were on the same oligo, the intended mutation rarely got in. It seemed that the cell was only using a part of the repair template and mutations further away from the cut site were not efficiently incorporated during HDR.
We decided to systematically characterize this phenomenon and found a general relationship between the distance of a mutation from the CRISPR/Cas9 cut site and how frequently it is incorporated. We were surprised to see how rapidly the efficiency of mutation incorporation falls with increasing distance from the cut site. If the distance is only 10 bp, the efficiency already drops by half and after only about 30 bp, it is no longer feasible to incorporate mutations without screening thousands of clones to find a positive one. So if you have better things to do than clone picking, choose your guide RNAs wisely (and let them cut close).
 |
| Figure 2: Relationship between cut site to mutation distance and editing efficiency. |
3. Optimizing "cut-to-mutation distance" for homo or heterozygous mutations
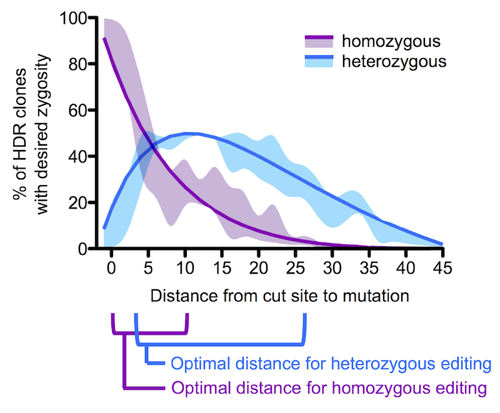
When thinking further about the distance relationship described above, we realized that, in addition to increasing mutation incorporation efficiency, we might exploit this relationship to direct the zygosity of the cells. This was important for us because the Alzheimer mutations we study are heterozygous in patients. As the probability of mutation incorporation drops with increasing cut-to-mutation distance, the mutation may only be inserted at one of the alleles, but not the other – the efficiency of editing is just too low for one to expect two successful mutations in a single cell.
We calculated the likelihood of obtaining homo- and heterozygous mutations by multiplying the single allelic mutation incorporation probabilities our data revealed, and found that for generating a homozygous HDR event with optimal efficiency a guide RNA targeting a cut <10 bp from the desired mutation should be used. For a heterozygous event, the cut should ideally be around 5 to 20 bp away from the cut site. Lastly, if a heterozygous event is desired but only guide RNAs that target very close to the intended mutation site are available, a repair template containing blocking and intended mutations can be equally mixed with another ssODN template that contains the same CRISPR/Cas-blocking mutation but not the intended mutation. Here, the blocking-only templates will compete with the blocking/intended mutation templates resulting in cells that use one or the other on each target site.
 |
| Figure 3: Optimal cut site to mutation distances for heterozygous and homozygous mutations. |
Taken together, for efficient and specific introduction of a desired sequence change by HDR, the optimal guide RNAs should not only have sufficient on-target and very low off-target activity, but should also mediate a DSB at optimal distance from the intended mutation site, depending on the desired zygosity. Furthermore, re-editing activity of the guide RNA should be minimized by incorporating CRISPR/Cas-blocking mutations into the HDR repair template.
Many thanks to our guest bloggers Dominik Paquet and Dylan Kwart
 Dominik Paquet is currently a Professor for Neurobiology at the Institute for Stroke and Dementia Research, Ludwig-Maximilians-University Munich. His lab develops iPSC-based models of neurodegenerative and neurovascular diseases. He worked on the described project while he was a Postdoc in Marc Tessier-Lavigne’s lab at the Rockefeller University in NYC.
Dominik Paquet is currently a Professor for Neurobiology at the Institute for Stroke and Dementia Research, Ludwig-Maximilians-University Munich. His lab develops iPSC-based models of neurodegenerative and neurovascular diseases. He worked on the described project while he was a Postdoc in Marc Tessier-Lavigne’s lab at the Rockefeller University in NYC.
 Dylan Kwart is a graduate student Marc Tessier-Lavigne’s lab at the Rockefeller University. He currently works to understand pathological mechanisms of Alzheimer’s disease by developing novel iPSC-based model systems.
Dylan Kwart is a graduate student Marc Tessier-Lavigne’s lab at the Rockefeller University. He currently works to understand pathological mechanisms of Alzheimer’s disease by developing novel iPSC-based model systems.
References
1. Cong, Le, et al. "Multiplex genome engineering using CRISPR/Cas systems." Science 339.6121 (2013): 819-823. PubMed PMID: 23287718. PubMed Central PMCID: PMC3795411.
2. Mali, Prashant, et al. "RNA-guided human genome engineering via Cas9." Science 339.6121 (2013): 823-826. PubMed PMID: 23287722. PubMed Central PMCID: PMC3712628.
3. Paquet, Dominik, et al. "Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9." Nature 533.7601 (2016): 125-125. PubMed PMID: 27120160.
4. Kwart, Dylan, et al. "Precise and efficient scarless genome editing in stem cells using CORRECT." Nature protocols 12.2 (2017): 329-354. PubMed PMID: 28102837.
Additional Resources on the Addgene Blog
- Learn about High Fidelity Cas9 Variants
- How to Design Your gRNA
- Delivering CRISPR Ribonucleoproteins
Resources on Addgene.org
- Browse All CRISPR Plasmids
- Check Out Our CRISPR Guide Pages
- Find Validated gRNAs

Topics: CRISPR, CRISPR Protocols and Tips





Leave a Comment