 Every quarter we highlight a subset of the new plasmids in the repository through our hot plasmids articles. These brief articles highlight the main features and applications of a partiular plasmid or set of plasmids. We hope that these articles make it easier for you to find and use the plasmids you need. You can find all the hot plasmids from 2016 below. With over 45,000 plasmids, we can't write posts for every great plasmid that comes into the repository, but be sure to let us know if you'd like to write about your plasmids in a future blog post. No time to read? Listen to our hot plasmids segment on the Addgene Podcast.
Every quarter we highlight a subset of the new plasmids in the repository through our hot plasmids articles. These brief articles highlight the main features and applications of a partiular plasmid or set of plasmids. We hope that these articles make it easier for you to find and use the plasmids you need. You can find all the hot plasmids from 2016 below. With over 45,000 plasmids, we can't write posts for every great plasmid that comes into the repository, but be sure to let us know if you'd like to write about your plasmids in a future blog post. No time to read? Listen to our hot plasmids segment on the Addgene Podcast.
View Plasmids from:
March (Q1) | June (Q2) | September 2016 (Q3) | December 2016 (Q3)
MARCH 2016
Enhancer activity mapping with STARR-seq

Although many technologies have been developed for the study of enhancer function, strength, and binding (e.g. DHS-seq, FAIRE-seq, and CHIP-seq), genome-wide identification and quantification of enhancer activity have remained challenging due to the variable availability of enhancer sequences in different chromatin states. Alexander Stark and colleagues at the Research Institute of Molecular Pathology have developed an alternative method, called STARR-seq (self-transcribing active regulatory region sequencing) that tests enhancer sequences outside of their endogenous chromatin environment and relies on the fact that enhancers function independently of their position relative to a transcription start site. In STARR-seq, randomly sheared genomic libraries are cloned downstream of a minimal promoter sequence. Should one of these random stretches of DNA contain an enhancer, it will activate its own transcription from the minimal promoter and its strength can be determined from its enrichment over input levels using next gen sequencing. The STARR-seq backbone is available from Addgene in both human and fly versions.
- Arnold, et al. Science. 2013. PubMed PMID: 23328393
Wu lab human lentiviral CRISPR library
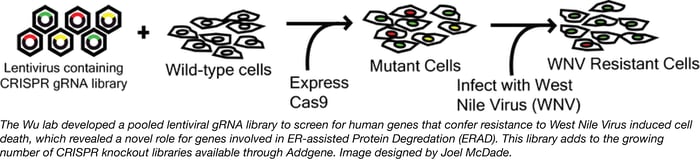
CRISPR pooled libraries enable researchers to screen the entire genome for genes that regulate a wide variety of phenotypes, including but not limited to cell growth and drug resistance. Recently, the Wu Lab at Texas Tech developed a pooled CRISPR knockout library and used this library to screen for genes involved in West Nile Virus (WNV) induced cell death.
The Wu lab library consists of 77,406 individual gRNAs targeting a total of 20,121 genes within the human genome (the identity of the genes and target sequences can be found here). To use the library, the authors packaged it into lentivirus and used the resulting lentiviral library to generate a population of HEK293FT cells expressing a single gRNA targeting a single gene. The gRNA expressing cells were then transfected with a Cas9-expressing plasmid to generate a population of mutant cells. The mutant cells were treated with WNV at a dose and duration sufficient to kill all control cells and surviving “mutant” colonies were deep sequenced to identify the gRNAs that confer protection to WNV. From this screen the authors were able to identify 7 previously unsuspected genes with known roles in ER-associated degradation (ERAD) that positively regulate the ability of WNV to induce cell death in infected cells (SEL1L, UBE2J1, EMC3, EMC2, DERL2, UBE2G2 and HRD1).
The Wu lab knockout library is available alongside a growing number of CRISPR libraries from Addgene’s repository that can be used to perform screens in human cells. General information regarding how to use a CRISPR pooled library as well as criteria of a good screening experiment can be found on our blog post entitled “Genome-wide Screening Using CRISPR/Cas9”. Detailed information regarding the Wu Lab library can be found in the associated publication.
- Ma, et al. Cell Rep. 2015. PubMed PMID: 26190106
PRESTO-TANGO: An open-source resource for drug discovery

Nearly one third of all medications target G-protein coupled receptors (GPCRs) yet one third of the GPCRs in the human genome are orphan receptors meaning their ligands are unknown. Identifying new ligands for orphan GPCRs is of significant medical interest, however efforts to do so have been hindered by a lack of tools, compounds, and assays to monitor activation of these receptors. Bryan Roth's lab has a newly available PRESTO-TANGO kit which allows researchers to interrogate the druggable human GPCR-ome using a quick, affordable, and ubiquitous reporter assay. In their recent publication, Kroeze et al. describe their enhanced TANGO arrestin recruitment assay incorporating 314 codon-optimized GPCR sequences for human cell line expression. In a twist on the classical TANGO assay, Kroeze et al. developed a new approach termed Parallel Receptor-ome Expression and Screening via Transcriptional Output-TANGO (PRESTO-TANGO) to screen the NCC-1 library of approved drugs against the entire kit; a parallel analysis that successfully identified new, highly specific agonists for orphan GPCRs demonstrating the utility of this platform for drug discovery. Researchers can use plasmids from the PRESTO-TANGO kit directly for drug screening or easily shuttle the optimized, expression validated GPCR sequences to any desired backbone.
- Kroeze, et al. Nat Struct Mol Biol. 2015. PubMed PMID: 25895059
- Barnea, et al. Proc Natl Acad Sci U S A. 2008. PubMed PMID: 18165312
CIDAR MoClo parts kit

Synthetic and basic biologists alike often need to test multiple expression systems before they achieve the appropriate expression level for their gene of interest. Although the parts involved in altering gene expression are relatively well known, particularly in bacteria, the process of cloning, combining, and testing these different parts can be quite arduous. The cloning process alone involves much literature searching, DNA synthesis, and plasmid assembly. Luckily, researchers from Douglas Densmore’s lab at Boston University have greatly simplified the process of generating multiple E. coli gene expression plasmids with the Cross-disciplinary Integration of Design Automation Research lab Moduclar Cloning (CIDAR MoClo) Parts Kit.
This Kit consists of a variety of plasmids containing many different promoters, RBS’, coding sequences (CDS), and terminators of varying strengths. These can be easily combined with your gene of interest and cloned into provided destination vectors using low-cost, one-pot golden gate cloning that results in the creation of many E. coli constructs with greatly varied levels of expression. The 93 plasmids in the CIDAR MoClo Parts Kit are available as glycerol stocks in a single plate with every plasmid or as individual plasmids shipped as agar stabs.
- Iverson, et al. ACS Synth Bio. 2015. PubMed PMID: 26479688.
RAS Pathway Clone Collection 2.0
 Ras proteins are small GTPases involved in cell signaling pathways that control many cellular processes such as differentiation, proliferation, and apoptosis. In humans, mutations that permanently activate Ras are found in one third of all cancers, including a high percentage (up to 95%) of pancreatic cancers. Ras oncogenes have been difficult to target therapeutically due in no small part to the central role Ras proteins play in cell signaling. Understanding how molecules in the Ras signaling pathway interact will allow scientists to develop better strategies for combating cancers. To facilitate innovative cancer research, the Reference Reagents Group of the NCI RAS Initiative, led by Dominic Esposito, has recently released a collection of 360 Ras pathway plasmids to the scientific community.
Ras proteins are small GTPases involved in cell signaling pathways that control many cellular processes such as differentiation, proliferation, and apoptosis. In humans, mutations that permanently activate Ras are found in one third of all cancers, including a high percentage (up to 95%) of pancreatic cancers. Ras oncogenes have been difficult to target therapeutically due in no small part to the central role Ras proteins play in cell signaling. Understanding how molecules in the Ras signaling pathway interact will allow scientists to develop better strategies for combating cancers. To facilitate innovative cancer research, the Reference Reagents Group of the NCI RAS Initiative, led by Dominic Esposito, has recently released a collection of 360 Ras pathway plasmids to the scientific community.
This collection consists of plasmids containing genes from a crowd-sourced map of the RAS signaling pathway and includes 180 unique transcripts. Each gene was chosen to represent the primary splice variant in 38 types of human cancer. Many of the transcripts are not available from other existing sources and are provided in both open (no stop codon) and closed (including a stop codon) formats. The collection is compatible with both standard Gateway cloning and the FNLCR Combinatorial Cloning Platform (CCP); the latter of which greatly enhances the utility of this collection as it can be used to easily create protein expression constructs fused with epitopes or fluorophores, generate expression vectors with various promoters suitable for in vivo expression, and/or produce vectors to make mutant cell lines or transgenic animals.
- Wall, et al. Methods Mol Biol. 2014. PubMed PMID: 24395366
Oxford Drosophila CRISPR library
 Addgene has had the privilege of distributing several human and murine CRISPR pooled libraries, but thanks to a deposit from the laboratory of Dr. Ji-Long Liu at Oxford University, we are now providing a powerful CRISPR-based screening source for the Drosophila community as well. The pooled library, designed to be used with cultured Drosophila cells, targets 13,501 fly genes with at least three independent gRNAs, each chosen strategically to maximize the likelihood of creating a functional knock-out via frameshift and minimize off-target effects. The library was cloned into the Liu lab’s own pAc-sgRNA-Cas9 insect expression backbone, which expresses the gRNA from a Drosophila U6:2 promoter and Cas9 from the actin 5C promoter. Addgene’s fly library web page offers more details and protocols, and the Liu lab’s OXfCRISPR page provides many other useful resources for fly labs who wish to harness this powerful genome engineering tool.
Addgene has had the privilege of distributing several human and murine CRISPR pooled libraries, but thanks to a deposit from the laboratory of Dr. Ji-Long Liu at Oxford University, we are now providing a powerful CRISPR-based screening source for the Drosophila community as well. The pooled library, designed to be used with cultured Drosophila cells, targets 13,501 fly genes with at least three independent gRNAs, each chosen strategically to maximize the likelihood of creating a functional knock-out via frameshift and minimize off-target effects. The library was cloned into the Liu lab’s own pAc-sgRNA-Cas9 insect expression backbone, which expresses the gRNA from a Drosophila U6:2 promoter and Cas9 from the actin 5C promoter. Addgene’s fly library web page offers more details and protocols, and the Liu lab’s OXfCRISPR page provides many other useful resources for fly labs who wish to harness this powerful genome engineering tool.
- Bassett, et al. J Genet Genomics. 2015. PubMed PMID: 26165496
GMAP - A new assembly platform for generating both simple and complex DNA constructs
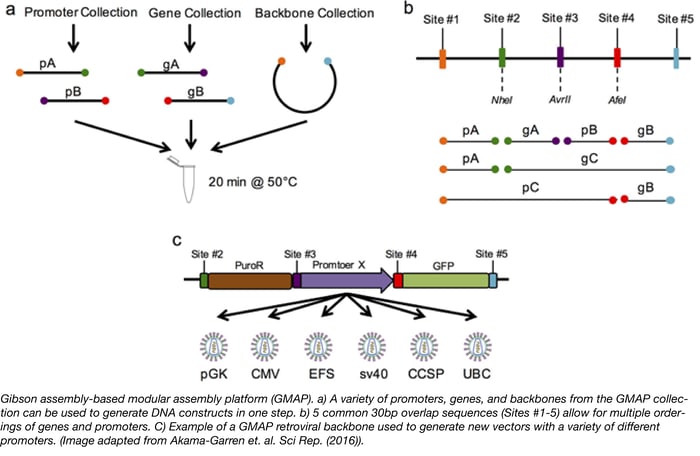
Have you ever wished you could design a gene knockdown or overexpression experiment, generate the necessary constructs, and complete screening in just 3 days? Thanks to Tyler Jacks’ lab, this 3-day goal is is now attainable. The Jacks lab has recently introduced a new assembly cloning platform called GMAP (Gibson assembly-based modular assembly platform). GMAP is based on the Gibson Assembly method, which allows for easy assembly of DNA fragments by utilizing regions of sequence homology (usually 30-40bp overlaps). To expand the capabilities of Gibson assembly and make a more modular platform, the Jacks lab generated 5 common 30bp overlap sequences (Sites #1-5). Each overlap site encodes a unique restriction enzyme site and unique primer sites for more rapid screening by restriction digest and Sanger sequencing, respectively. The speed and simplicity of GMAP allows scientists to easily design, generate, and test constructs composed of complex genetic elements.
To demonstrate the usefulness of GAMP, the Jacks Lab constructed several GMAP-compatible vectors for a wide variety of biological applications. First, GMAP-compatible backbones for lentivirus LV 1-5 and retrovirus RV 2-5 were constructed. The authors then used GMAP assembly to establish a collection of over 30 promoters and 140 genes, including constructs with unique tissue-specific promoters expressing GFP, tetracycline response elements, and shRNAs many of which are available at Addgene. Since GMAP assembly utilizes 5 different overlap sites, this technique also allows for easy and rapid targeting of complex elements to specific genomic sites. For instance, the authors generated a GMAP-compatible backbone containing Rosa26 homology arms. The Rosa26 locus is commonly used to drive ubiquitous expression of a target gene in mice. The Jacks Lab used this backbone to target a CAG-driven loxP-stop-loxP cassette into the Rosa26 locus, allowing for Cre-dependent expression of a GMAP inserted gene. The applications for such a modular system are endless and, as scientists use GMAP, there will be a continuous expansion of compatible backbones, promoters, and genes available to the community. If you plan to use GMAP for your experiments, think about depositing your new plasmids with Addgene to help expand the collection!
The Jacks lab has made several GMAP-compatible plasmids available to the scientific community through Addgene.
- Akama-Garren, et al. J Genet Genomics. 2016. PubMed PMID: 26887506
Toronto KnockOut (TKO) CRISPR library
 In order to identify both core and context-dependent human fitness genes, the laboratory of Jason Moffat at the University of Toronto has created the Toronto KnockOut (TKO) CRISPR Library. This complex second-generation CRISPR lentiviral library targets nearly all human protein-coding genes, and is unique in that it is composed of two sub-pools. The first is a base “90k library” that contains ~90,000 gRNAs that target ~15,000 genes with 6 gRNAs per gene. The second is a "supplementary" library that contains an additional 6 gRNAs per gene generated using slightly relaxed gRNA design parameters.
In order to identify both core and context-dependent human fitness genes, the laboratory of Jason Moffat at the University of Toronto has created the Toronto KnockOut (TKO) CRISPR Library. This complex second-generation CRISPR lentiviral library targets nearly all human protein-coding genes, and is unique in that it is composed of two sub-pools. The first is a base “90k library” that contains ~90,000 gRNAs that target ~15,000 genes with 6 gRNAs per gene. The second is a "supplementary" library that contains an additional 6 gRNAs per gene generated using slightly relaxed gRNA design parameters.
To demonstrate the effectiveness of their library design, Moffat laboratory members conducted multiple screens in different human cell models and identified ~4-5-fold more fitness-related genes than have been found in previous screens of this type. They were also able to functionally characterize positive hits. This work has been published in Cell.
The Moffat laboratory maintains a website for the TKO library that contains up-to-date information and data-sets, as well as an interactive gRNA viewer. The Moffat lab has also deposited their Cas9 and gRNA backbone plasmids with Addgene. Additional control plasmids for the TKO library will be available at Addgene soon.
- Hart, et al. Cell. 2015. PubMed PMID: 26627737
pORTMAGE: A portable method for bacterial genome engineering across multiple species
 Recent discoveries in the fields of genome engineering and synthetic biology have transformed the way scientists create new traits in bacteria and enabled the in-depth study of many biological processes. While these methods can be used to easily modify an individual locus within the genome, they tend to show their limits when it comes to simultaneously modifying multiple loci (multiplexing).
Recent discoveries in the fields of genome engineering and synthetic biology have transformed the way scientists create new traits in bacteria and enabled the in-depth study of many biological processes. While these methods can be used to easily modify an individual locus within the genome, they tend to show their limits when it comes to simultaneously modifying multiple loci (multiplexing).
Currently, the only genome engineering method in bacteria that enables rapid, automated, and high-throughput genome editing is multiplex automated genome engineering (MAGE) (1). MAGE uses recombineering (2) to simultaneously incorporate multiple single-strand DNA (ssDNA) oligonucleotides (oligos), and thereby rapidly create desired allele combinations and combinatorial genomic libraries. MAGE has allowed genome-engineering endeavours of unparalleled complexity in Escherichia coli, like the construction of a so-called “genomically recoded organism” (3) but its portability to other bacterial strains remains seriously limited, as prior optimizations are required for each individual target species. Indeed, in order to use MAGE, the λ Red recombinase enzymes need to be expressed and the native methyl-directed mismatch repair (MMR) system needs to be repressed in the host strain.
To address this problem, Csaba Pál’s lab has created a set of vectors that allows one to use the MAGE method in unmodified bacterial species (4). This set of plasmids (dubbed pORTMAGE) expresses the λ Red recombinase enzymes, as well as a dominant-negative mutator allele of the E. coli MMR protein MutL, all under the control of the cI857 temperature-sensitive repressor. The temperature-controlled expression of the MutL mutant enables transient suppression of DNA repair during oligonucleotide integration, allowing MAGE in otherwise unmodified bacterial strains (5). MutL is highly conserved in distant relatives of E. coli so it can be used in a broad range of strains. Thus, pORTMAGE simultaneously allows genome editing and mutant library generation in several biotechnologically and clinically relevant bacterial species.
pORTMAGE vectors open new horizons to modify genomes in a broad range of bacterial hosts. This fantastic tool is now available at Addgene so you can be the “MAGE” that will create new traits in bacteria.
-
Wang, et al. Nature. 2009. PubMed PMID: 19633652
-
Court, et al. Annu Rev Genet. 2002. PubMed PMID: 12429697
-
Lajoie, et al. Science. 2013. PubMed PMID: 24136966
-
Nyerges, et al. Proc Natl Acad Sci USA. 2016. PubMed PMID: 26884157
-
Aronshtam, et al. Nucleic Acids Res. 1996. PubMed PMID: 8692687
Tracking autophagy in yeast with Rosella, a pH-sensitive fluorescent biosensor
 A pH-sensitive fluorescent biosensor-based assay system was recently deposited with Addgene by Dr. Mark Prescott. Dr. Prescott and Dr. Rod Devenish (both at Monash University) originally developed the system for monitoring and analysing autophagy of cytosol and organelles in yeast cells. Named after the brightly-coloured Australian parrot, the dual colour-emission biosensor Rosella consists of DsRed connected to a pH-sensitive variant of GFP (SEP) by a 9 amino-acid linker (see Figure 1). The key to the biosensor lies in pH: DsRed is relatively pH-insensitive, while SEP fluoresces at cellular pH (7.0) but not at vacuolar pH (6.2). The colour of Rosella therefore varies with cellular location: it is both red and green through much of the cell, but turns more red in lower (e.g. vacuolar) pH environments that inactivate SEP and leave only DsRed to fluoresce (1). With variants that can be targeted to specific cellular compartments (cytosol, Addgene catalog #71245; mitochondria, Addgene catalog #71247) Rosella provides an easy way to follow uptake of cellular material into the yeast vacuole, as well as to examine the mechanisms behind autophagic pathways.
A pH-sensitive fluorescent biosensor-based assay system was recently deposited with Addgene by Dr. Mark Prescott. Dr. Prescott and Dr. Rod Devenish (both at Monash University) originally developed the system for monitoring and analysing autophagy of cytosol and organelles in yeast cells. Named after the brightly-coloured Australian parrot, the dual colour-emission biosensor Rosella consists of DsRed connected to a pH-sensitive variant of GFP (SEP) by a 9 amino-acid linker (see Figure 1). The key to the biosensor lies in pH: DsRed is relatively pH-insensitive, while SEP fluoresces at cellular pH (7.0) but not at vacuolar pH (6.2). The colour of Rosella therefore varies with cellular location: it is both red and green through much of the cell, but turns more red in lower (e.g. vacuolar) pH environments that inactivate SEP and leave only DsRed to fluoresce (1). With variants that can be targeted to specific cellular compartments (cytosol, Addgene catalog #71245; mitochondria, Addgene catalog #71247) Rosella provides an easy way to follow uptake of cellular material into the yeast vacuole, as well as to examine the mechanisms behind autophagic pathways.
- Rosado, et al. Autophagy. 2008. PubMed PMID: 18094608
JUNE 2016
Repurposing the CRISPR-Cas9 system for targeted DNA methylation
 The traditional approach to studying epigenetic modifications has been to use inhibitors of epigenetic modifiers like DNA methyltransferases or histone deacetylases. Unfortunately, these inhibitors are often non-selective, affecting the entire genome and not specific loci. To overcome this flaw, the Zoldos lab has developed CRISPR tools for epigenome editing that enable the precise study of particular epigenetic modifications and their effects on gene regulation.
The traditional approach to studying epigenetic modifications has been to use inhibitors of epigenetic modifiers like DNA methyltransferases or histone deacetylases. Unfortunately, these inhibitors are often non-selective, affecting the entire genome and not specific loci. To overcome this flaw, the Zoldos lab has developed CRISPR tools for epigenome editing that enable the precise study of particular epigenetic modifications and their effects on gene regulation.
Professor Zoldos and colleagues from the Department of the Biology, Division of Molecular Biology at the University of Zagreb, constructed a CRISPR-Cas9-based tool for targeting CpG methylation by fusing catalytically inactive Cas9 (dCas9) with the catalytic domain from DNA methyltransferase, DNMT3A. Co-expression of this modified Cas9 along with a gRNA targeting a specific genomic locus causes that locus to be methylated. This powerful tool has been used to direct methylation to and lower expression from both the IL6ST and BACH2 promoters.
- Vojta A, et al. Nucl. Acids Res. 2016. PubMed PMID: 26969735
Updated biotinylation tools to identify proximate proteins
 The Roux Lab has deposited a smaller version of the promiscuous biotin ligase used in their popular biotinylation method for identifying protein-protein associations. This method uses a bait protein fused to a biotin ligase to biotinylate proximate proteins in the cell. These candidate proteins can be enriched via biotin pull-down methods and identified using mass spectrometry.
The Roux Lab has deposited a smaller version of the promiscuous biotin ligase used in their popular biotinylation method for identifying protein-protein associations. This method uses a bait protein fused to a biotin ligase to biotinylate proximate proteins in the cell. These candidate proteins can be enriched via biotin pull-down methods and identified using mass spectrometry.
The Roux Lab’s new and improved version of BioID, called BioID2, is significantly smaller, allows for more selective targeting of fusion proteins, requires less biotin, and shows enhanced labeling of proximate proteins. Overall BioID2 improves the efficiency of screening for protein–protein interactions.
The Roux lab has deposited HA tagged BioID2 for N-terminal fusions and Myc tagged BioID2 for C-terminal fusions.
- Kim, et al. Mol Biol Cell. 2016. PubMed PMID: 26912792
FusX-TALEN assembly system
 TALEs as nucleases (TALENs) are genome-editing tools that are deployed in both in vitro cell systems and diverse model organisms. Addgene’s newest TALEN kit, the FusX assembly system from Stephen Ekker’s lab, is a modified version of the Golden Gate TALEN kit (GGT) that streamlines the assembly of TALE repeats into a single-tube, 3 day process by utilizing pre-assembled trimers to reduce the number of cloning steps. The FusX system was verified in zebrafish and was shown to offer high activity and unparalleled specificity in genomic targeting design flexibility. The FusX kit is backward compatible with all TAL effector scaffolds previously constructed for the Golden Gate TALEN and TAL Effector Kit 2.0.
TALEs as nucleases (TALENs) are genome-editing tools that are deployed in both in vitro cell systems and diverse model organisms. Addgene’s newest TALEN kit, the FusX assembly system from Stephen Ekker’s lab, is a modified version of the Golden Gate TALEN kit (GGT) that streamlines the assembly of TALE repeats into a single-tube, 3 day process by utilizing pre-assembled trimers to reduce the number of cloning steps. The FusX system was verified in zebrafish and was shown to offer high activity and unparalleled specificity in genomic targeting design flexibility. The FusX kit is backward compatible with all TAL effector scaffolds previously constructed for the Golden Gate TALEN and TAL Effector Kit 2.0.
Ma, et al. Hum Gene Ther. 2015. PubMed PMID: 26854857
Examples of FusX-Compatible Destination Vectors Available at Addgene:
| Lab | Plasmids |
| Dan Carlson | RCIscript-GoldyTALEN, pC-GoldyTALEN |
| Tom Ellis | pTAL5-BB, pTAL6-BB |
| David Grunwald | pCS2TAL3-DD, pCS2TAL3-RR |
| Pawel Pelczar | pCAG-T7-TALEN(Sangamo)-Destination series, pCAG-Golden-Gate-Esp3I-Destination |
| Takashi Yamamoto | pcDNA-TAL-NC2, pCAGGS-TAL-NC2 |
| Charles Gersbach | pcDNA3.1-GoldenGate, pcDNA3.1-GoldenGate-VP64 |
| Maria-Elena Torres-Padilla | pTALYM3, pTALYM4 |
| Boris Greber | pTAL7a, pTAL7b |
| Michal Zurovec | pBlue-TAL |
| Nathan Lawson | pJDS Series |
Superglue proteins with SpyTag/SpyCatcher and SnoopTag/SnoopCatcher
 The development of recombinant DNA technology has allowed molecular biologists to fuse fragments of DNA together in a desired order and express fusion proteins in a target cell. In addition to nucleotide-based linking strategies, post translational protein fusion would be advantageous when individual protein expression is preferred or required, or when it is difficult for a cell to synthesize or fold a protein because of its length. Mark Howarth’s lab recently engineered a new peptide/protein pair, SnoopTag/SnoopCatcher, that can be used to irreversibly link desired proteins together via a spontaneous isopeptide bond. When used in combination with their previously described SpyTag/SpyCatcher pair, multiple protein fusions can be synthesized in an iterative process by alternating the use of the SnoopTag/SnoopCatcher and SpyTag/SpyCatcher pairs.
The development of recombinant DNA technology has allowed molecular biologists to fuse fragments of DNA together in a desired order and express fusion proteins in a target cell. In addition to nucleotide-based linking strategies, post translational protein fusion would be advantageous when individual protein expression is preferred or required, or when it is difficult for a cell to synthesize or fold a protein because of its length. Mark Howarth’s lab recently engineered a new peptide/protein pair, SnoopTag/SnoopCatcher, that can be used to irreversibly link desired proteins together via a spontaneous isopeptide bond. When used in combination with their previously described SpyTag/SpyCatcher pair, multiple protein fusions can be synthesized in an iterative process by alternating the use of the SnoopTag/SnoopCatcher and SpyTag/SpyCatcher pairs.
In order to improve the ease and convenience of assembling these multi-protein fusions or “polyproteams”, the Howarth lab created the MBPx-SpyCatcher construct. This construct is used to produce a maltose binding protein/SpyCatcher fusion that can be anchored to amylose resin for solid-phase synthesis of larger fusions through SpyTag/SpyCatcher and SnoopTag/SnoopCatcher linkages. Once all of the desired sequential additions have been made, the final polyproteam can be eluted using maltose. This procedure allows for stable and directional protein fusion, with a SpyCatcher-SnoopCatcher linker remaining between each protein unit, and provides a new method for generating protein chains or clusters.
Veggiani, et al. Proc Natl Acad Sci U S A. 2016. PubMed PMID: 26787909
EasyClone 2.0 Yeast Toolkit
 Many industries, such as the food and beverage industry, the bioethanol industry, and the pharmaceutical industry, depend on the yeast, Saccharomyces cerevisiae, as a key host for the production of their products, including fermented beer and wine, fuels, pharmaceutical ingredients, and recombinant proteins. In order to keep production yields high and costs low, and to deal with the increasing variety of new compounds being generated, there is a need for genetic optimization of industrial yeast strains. However, industrial yeast strains are more difficult to genetically modify than common lab yeast strains since they are prototrophic, usually have low levels of homologous recombination, and can be difficult to transform.
Many industries, such as the food and beverage industry, the bioethanol industry, and the pharmaceutical industry, depend on the yeast, Saccharomyces cerevisiae, as a key host for the production of their products, including fermented beer and wine, fuels, pharmaceutical ingredients, and recombinant proteins. In order to keep production yields high and costs low, and to deal with the increasing variety of new compounds being generated, there is a need for genetic optimization of industrial yeast strains. However, industrial yeast strains are more difficult to genetically modify than common lab yeast strains since they are prototrophic, usually have low levels of homologous recombination, and can be difficult to transform.
To increase the genetic toolbox for industrial yeast strains, the Borodina Lab has recently designed the EasyClone 2.0 toolkit. EasyClone 2.0 consists of 25 integrative vectors containing long homology arms and both auxotrophic and dominant selection markers, allowing for use with both laboratory and prototrophic strains. The vectors were based on the original EasyClone vectors (Jensen et al 2013) allow for cloning of up to two genes with a bidirectional promoter, integrate at 11 specific chromosomal locations, and stably express the integrated gene. These vectors contain uracil-excision based (USER) sites, flanked by ADH1 and CYC terminators for easy cloning of genes and promoters. In addition to USER cloning, this toolkit is also compatible with systems such as in-fusion, Gibson, and MoClo. The EasyClone 2.0 kit has been made publically available through Addgene.
- Stovicek, et al. J Ind Microbiol Biotechnol. 2015. PubMed PMID: 26376869
- Jensen, et al. FEMS Yeast Research. 2014. PubMed PMID: 24151867
- Mikkelson, et al. Metab Eng. 2012. PubMed PMID: 2232647
SapTrap, a toolkit for high-throughput CRISPR/Cas9 gene modification in Caenorhabditis elegans
 CRISPR/Cas9 technology has revolutionized genome editing in multiple organisms thanks to its simple, easily modifiable RNA-guided targeting mechanism. However, the laborious construction of repair templates and guide RNA constructs, as well as the difficult screens required to identify correctly modified organisms has limited routine use of this technology. To streamline CRISPR/Cas9-based genetic tag insertion in the C. elegans genome, Erik Jorgensen’s lab from the University of Utah developed SapTrap.
CRISPR/Cas9 technology has revolutionized genome editing in multiple organisms thanks to its simple, easily modifiable RNA-guided targeting mechanism. However, the laborious construction of repair templates and guide RNA constructs, as well as the difficult screens required to identify correctly modified organisms has limited routine use of this technology. To streamline CRISPR/Cas9-based genetic tag insertion in the C. elegans genome, Erik Jorgensen’s lab from the University of Utah developed SapTrap.
The SapTrap system uses golden gate assembly to produce single plasmid targeting vectors that encode both a guide RNA transcript and a repair template for an individual tagging event. Commonly used sequences, including fluorescent tags, a floxed Cbr-unc-119 selectable marker, and “connectors” that link tags to the targeted gene are supplied to the reaction from a prebuilt donor plasmid library, available from Addgene. Site-specific sequences for homology arms and the guide RNA are supplied as annealed synthetic oligos - eliminating the need for PCR or additional molecular cloning steps during plasmid assembly.
This toolkit reduces the expense and workload necessary to produce vectors for genome editing in worms to the point that high-throughput tagging projects can be performed.
Schwartz, et al. Genetics. 2016. PubMed PMID: 26837755
SEPTEMBER 2016
Zika virus plasmids Now avaliable through Addgene
 The Vaithi Arumugaswami lab at Cedars-Sinai Medical Center has kindly constructed and deposited an unpublished set of Zika gene plasmids to Addgene’s repository. The genes, from the Zika virus Asian genotype PRVABC59, were obtained from the CDC and cloned into a third generation lentiviral transfer vector. Each gene has been FLAG-tagged for convenient experimental use. The plasmids were validated by both Western blotting and immunohistochemistry, and are summarized in the table below:
The Vaithi Arumugaswami lab at Cedars-Sinai Medical Center has kindly constructed and deposited an unpublished set of Zika gene plasmids to Addgene’s repository. The genes, from the Zika virus Asian genotype PRVABC59, were obtained from the CDC and cloned into a third generation lentiviral transfer vector. Each gene has been FLAG-tagged for convenient experimental use. The plasmids were validated by both Western blotting and immunohistochemistry, and are summarized in the table below:
| ID | Cv (Capsid) | FLAG Tag Terminus |
| 79629 (N) and 79628 (C) | Cv (capsid) | N and C |
| 79633 (N) and 79641 (C) | NS1 (var: W98G) | N and C |
| 79635 | NS3 | C |
| 79637 | NS2B | C |
| 79636 | NS4A | N |
| 79640 | NS4B | C |
| 79639 | NS5 | C |
| 79632 (N) and 79631 (C) | PrM | N and C |
Detecting protein-protein interactions with DULIP
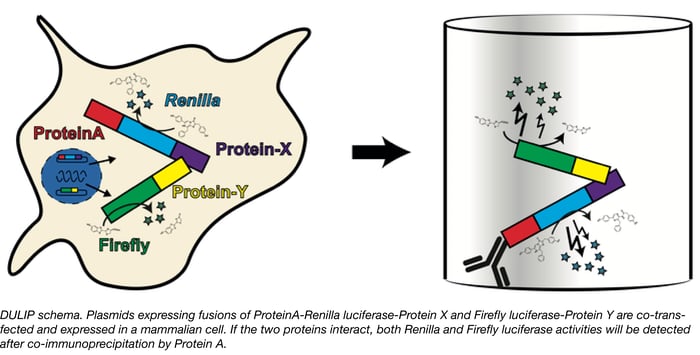
Proteins rarely act alone. Molecular processes from signal transduction to transport rely on protein-protein interactions and therefore identifying and studying interaction networks is important to detemine how these systems work. Researchers screening protein-protein interactions in mammalian systems have many assays to choose from, however, these methods are not without limitations. Some, such as the well-known yeast two-hybrid assay are not optimal for studying native mammalian protein interactions, whereas others may be too cumbersome to use in a high throughput manner or make it difficult to quantify interaction strengths. Erich Wanker’s group at the Max-Delbrueck Center for Molecular Medicine has recently developed a new method called DULIP (dual luminescence-based co-immunoprecipitation) to overcome the limitations of other methods and allow researchers to robustly identify and quantify protein-protein interactions in mammalian systems.
The DULIP system is comprised of Gateway-compatible plasmids in which “bait” proteins are tagged with Protein A-Renilla luciferase and the “prey” proteins are tagged with Firefly luciferase. The bait and prey plasmids are co-transfected and proteins are immunoprecipitated from the cells using the Protein A tag. The luciferase activities can then be measured and normalized to assess interactions. Higher relative firefly luciferase activity in the Co-IP compared of a control Co-IP indicates stronger interaction. The system has been demonstrated to detect high and low affinity interactions, identify changes in interaction strength due to point mutations, and is suitable for high throughput applications.
- Trepte P, et al. J. Mol. Biol. 2015. PubMed PMID: 26264872
Rapid, conditional degradation of essential proteins using Cas9-based AID tagging
 Seven years ago, Masato Kanemaki's lab successfully used the plant-based auxin-inducible degron (AID) technology to conditionally knock down proteins in a variety of model organisms. As long as it was expressed in a cell that also expressed auxin-sensitive TIR1, any protein fused with their AID tag could be targeted for degredation via ubiquitination. The key feature of auxin-induced degredation is the speed at which it works--typical half lives of AID-tagged proteins after the introduction of auxin range from 10-20 minutes, making AID an appealing tool for studying essential proteins.
Seven years ago, Masato Kanemaki's lab successfully used the plant-based auxin-inducible degron (AID) technology to conditionally knock down proteins in a variety of model organisms. As long as it was expressed in a cell that also expressed auxin-sensitive TIR1, any protein fused with their AID tag could be targeted for degredation via ubiquitination. The key feature of auxin-induced degredation is the speed at which it works--typical half lives of AID-tagged proteins after the introduction of auxin range from 10-20 minutes, making AID an appealing tool for studying essential proteins.
Until recently, it was challenging to apply the AID-tagging system to essential genes in human cells and mouse ES cells because it was difficult to fuse the AID tag to endogenous proteins. As described in their recent Cell Reports paper, the Kanemaki lab overcame this hurdle using CRISPR technology. First, they used CRISPR to create OsTIR1-expressing cell lines by inserting OsTIR1 into the "safe harbor" AAVS1 locus (a tet-inducible OsTIR1 plasmid is also available). Second, they co-transfected short-arm donor vectors containing mAID and either Neo or Hygro resistance markers with CRISPR/Cas vectors targeting their genes of interest to generate mAID fusions to endogenous genes. Cell lines with the appropriate fusions could then be selected for using the indicated antibiotics. In addition, some versions of the mAID vectors come with mCherry2 or mClover fluorescent proteins allowing you to screen for fusions. This combination of CRISPR and AID technologies provides a clever means of investigating hard-to-study genes in hard-to-study cell types.
- Natsume T, et al. Cell Rep. 2016. PubMed PMID: 27052166
- Nishimura K, et al. Nat. Methods 2009. PubMed PMID: 19915560
TRIBE: Targets of RNA-binding proteins identified by editing
 Identifying targets of RNA-binding proteins (RBPs) has traditionally been difficult, as techniques like CLIP require a specific antibody and a large number of cells to analyze. A new technique from Michael Rosbash’s lab, TRIBE, solves both of these problems by fusing an RBP to the catalytic domain of RNA editing enzyme, ADAR. This RBP-ADARcd fusion protein irreversibly edits adenine residues in the region of mRNA binding, and these sequence changes are subsequently identified through transcriptome sequencing. Importantly, results obtained using TRIBE compared favorably to those of CLIP, the gold standard for characterizing RBP targets.
Identifying targets of RNA-binding proteins (RBPs) has traditionally been difficult, as techniques like CLIP require a specific antibody and a large number of cells to analyze. A new technique from Michael Rosbash’s lab, TRIBE, solves both of these problems by fusing an RBP to the catalytic domain of RNA editing enzyme, ADAR. This RBP-ADARcd fusion protein irreversibly edits adenine residues in the region of mRNA binding, and these sequence changes are subsequently identified through transcriptome sequencing. Importantly, results obtained using TRIBE compared favorably to those of CLIP, the gold standard for characterizing RBP targets.
One key advantage of TRIBE is the low input required - McMahon et al., report using as few as 150 fly neurons as their starting material! Thus, TRIBE can be used to analyze a particular cell population within a given tissue rather than a mixed sample. TRIBE was developed in Drosophila, and further work is needed to assess its function in mammalian cells or other animal models, but this technique has laid the groundwork for better analysis of RBP function.
- McMahon A, et al. Cell. 2016. PubMed PMID: 27040499
PiggyBac mediated delivery of robust CRISPR activators
 We previously reported on a novel Cas9-based activator developed by the Church lab, termed “dCas9-VPR”, which facilitates robust activation of target genes (20-40x greater than standard dCas9-VP64 activators) when introduced into mammalian cells.
We previously reported on a novel Cas9-based activator developed by the Church lab, termed “dCas9-VPR”, which facilitates robust activation of target genes (20-40x greater than standard dCas9-VP64 activators) when introduced into mammalian cells.
Although the VPR activation domain is an incredible tool for researchers trying to activate target genes, the relatively large size of dCas9-VPR limits one’s ability to package it into lentivirus and later create stable cell lines expressing it. The Church lab fixed this issue by creating a new dCas9-VPR expression plasmid that uses the piggybac transposase to integrate dCas9-VPR into the genome of target cells. The piggybac system has a much larger cargo capacity than lentivirus and is capable of efficiently creating stable cell lines using direct transfection precluding the need for virus. Furthermore, dCAS9-VPR expression is under the control of the TRE promoter, which allows for doxycycline-inducible expression. The end result is genomic integration of dCas9-VPR and controlled expression of the dCas9-VPR transgene by doxycycline in your target cells.
This plasmid adds to the growing collection of dCas9-VPR plasmids, which already includes dSaCas9-VPR in an AAV transfer vector for in-vivo expression and plasmids optimized for dCas9-VPR expression in drosophila and yeast.
- Chavez A, et al. Nat. Methods 2015. PubMed PMID: 25730490
New kit for editing dicotyledonous plants
 The Stuttmann lab generated the vectors in this kit to make it easy to apply a variety of CRISPR tools to dicotyledonous plants. The pDGE Dicot Genome Editing Kit contains Cas9, Cas9 nickase, and Cas9 activator expressing plasmids that can co-express 1-8 gRNAs. Ordon et al., 2016 tested the Cas9 constructs for their ability to generate both small (< 100 bp) and large (up to 120 kb) deletions in N. benthamiana and Arabidopsis. The authors found that deletion efficiency was dependent upon 1) Cas9 and gRNA dose (both of which can be modulated using the different promoters in the kit and by altering the number of tandemly expressed gRNAs respectively) and 2) the size of the deletion. Smaller deletions generally occurred at higher frequency and could be detected by PCR while large deletions were relatively rare. The vectors are compatible with Golden Gate Cloning and Agrobacterium-mediated delivery making them easy to customize for targeting your gene of interest. Try them today!
The Stuttmann lab generated the vectors in this kit to make it easy to apply a variety of CRISPR tools to dicotyledonous plants. The pDGE Dicot Genome Editing Kit contains Cas9, Cas9 nickase, and Cas9 activator expressing plasmids that can co-express 1-8 gRNAs. Ordon et al., 2016 tested the Cas9 constructs for their ability to generate both small (< 100 bp) and large (up to 120 kb) deletions in N. benthamiana and Arabidopsis. The authors found that deletion efficiency was dependent upon 1) Cas9 and gRNA dose (both of which can be modulated using the different promoters in the kit and by altering the number of tandemly expressed gRNAs respectively) and 2) the size of the deletion. Smaller deletions generally occurred at higher frequency and could be detected by PCR while large deletions were relatively rare. The vectors are compatible with Golden Gate Cloning and Agrobacterium-mediated delivery making them easy to customize for targeting your gene of interest. Try them today!
- Ordon J, et al. Plant J. 2016. PubMed PMID: 27579989
DECEMBER 2016
Restoring yeast prototrophy with plasmids
 Though auxotrophic markers are used frequently for selection in yeast, Addgene's own Plasmids 101: Yeast Vectors blog post concedes that there are some drawbacks with this technique. Great care needs to be taken with common S288c strains since failing to fully complement strains with multiple auxotrophies or using different auxotrophic backgrounds can lead to disparate physiological effects and complicate any metabolic study. The Ralser lab has created a unique tool with their Yeast Prototrophy Kit, a set of plasmids designed to complement unused auxotrophies in Saccharomyces strains by compensating for histidine (HIS3), leucine (LEU2), uracil (URA3), methionine (MET17), and lysine (LYS2) deficiencies, and combinations thereof. The 23 plasmids in this kit are derived from the Ralser lab's own pHLUM minichromosomal vector, and besides being used for restoring strain prototrophy, can also aid in designing self-establishing metabolically cooperating (SeMeCo) communities. The uniform multiple cloning site in the plasmid series also allows for protein expression under a range of auxotrophic markers. Read more about the Ralser lab's important new tool for the yeast community in their open access article.
Though auxotrophic markers are used frequently for selection in yeast, Addgene's own Plasmids 101: Yeast Vectors blog post concedes that there are some drawbacks with this technique. Great care needs to be taken with common S288c strains since failing to fully complement strains with multiple auxotrophies or using different auxotrophic backgrounds can lead to disparate physiological effects and complicate any metabolic study. The Ralser lab has created a unique tool with their Yeast Prototrophy Kit, a set of plasmids designed to complement unused auxotrophies in Saccharomyces strains by compensating for histidine (HIS3), leucine (LEU2), uracil (URA3), methionine (MET17), and lysine (LYS2) deficiencies, and combinations thereof. The 23 plasmids in this kit are derived from the Ralser lab's own pHLUM minichromosomal vector, and besides being used for restoring strain prototrophy, can also aid in designing self-establishing metabolically cooperating (SeMeCo) communities. The uniform multiple cloning site in the plasmid series also allows for protein expression under a range of auxotrophic markers. Read more about the Ralser lab's important new tool for the yeast community in their open access article.
- Mülleder M, et al. F1000Res. PubMed PMID: 27830062
Toxoplasma CRISPR knockout pooled library
 Parasites from the apicomplexan phylum cause severe human and livestock diseases such as malaria and toxoplasmosis. Despite their importance to global health, most apicomplexan genes remain uncharacterized. Recent studies carried out by Sebastian Lourido and colleagues constitute an important step toward understanding the biology of these parasites. In this work, they used the CRISPR/Cas9 technology to measure the contribution of each gene from the model apicomplexan organism Toxoplasma gondii to the infection of human fibroblasts.
Parasites from the apicomplexan phylum cause severe human and livestock diseases such as malaria and toxoplasmosis. Despite their importance to global health, most apicomplexan genes remain uncharacterized. Recent studies carried out by Sebastian Lourido and colleagues constitute an important step toward understanding the biology of these parasites. In this work, they used the CRISPR/Cas9 technology to measure the contribution of each gene from the model apicomplexan organism Toxoplasma gondii to the infection of human fibroblasts.
Toxoplasma mutants were generated by transforming parasites that constitutively express Cas9 with a library of sgRNA expression vectors (containing ten guides against each of the 8,158 predicted T. gondii protein-coding genes). Dr. Lourido’s studies demonstrate that genome-scale genetic screening is an efficient approach to identify genes involved in drug-resistance. Remarkably, this method also enabled Lourido’s group to define ~200 previously uncharacterized genes that are conserved among apicomplexans and important for T. gondii fitness. Sixteen of these genes were investigated for functions during infection of human cells, and one of them called “claudin-like apicomplexan microneme protein” (CLAMP) was shown to be important for the initiation of the T. gondii infection, and necessary for the asexual cycle of malaria parasites.
Find Lourido CRISPR Plasmids.
- Sidik and Huet, et al. Cell. 2016. PubMed PMID: 27594426
CRISPR-X: dCas9-targeted point mutations for directed evolution
 Directed evolution is an important method for protein engineering, but it can be difficult to generate enough variation in a native genomic context to find useful mutants. CRISPR-X, a new system from Michael Bassik’s lab, overcomes this obstacle. With CRISPR-X, dCas9 is used to recruit an sgRNA with MS2 hairpin binding sites, which then recruit an MS2-fused hyperactive variant of activation-induced cytidine deaminase (AID).
Directed evolution is an important method for protein engineering, but it can be difficult to generate enough variation in a native genomic context to find useful mutants. CRISPR-X, a new system from Michael Bassik’s lab, overcomes this obstacle. With CRISPR-X, dCas9 is used to recruit an sgRNA with MS2 hairpin binding sites, which then recruit an MS2-fused hyperactive variant of activation-induced cytidine deaminase (AID).
AID normally mediates somatic hypermutation to generate diverse antibodies; in the CRISPR-X system, AID*Δ similarly produces single base changes at each gRNA-specified target locus. Within the editing window -50 to +50 bp from the PAM site, CRISPR-X produces point mutations at a rate of up to 1 per 500-1000 bp. In comparison, the DNA replication error rate is 1 per 109 bp.
Unlike wt Cas9, CRISPR-X produces few indels, so it maintains reading frame and instead creates diverse, localized point mutations. As a proof of concept, Hess et al. successfully evolved wild type GFP to EGFP using CRISPR-X and subsequent FACS sorting. They also mutagenized proteasome subunit PSMB5 to find mechanisms of resistance to a proteasome inhibitor; CRISPR-X identified both previously characterized and novel mutants. Hess et al. envision CRISPR-X as not only a protein engineering tool, but also as a way to characterize/engineer promoters, enhancers, and other regulatory elements.
Find the CRISPR-X Plasmids
- Hess G, et al. Nat. Methods 2016. PubMed PMID: 27798611
Magneto 2.0 - A magnetic remote control for the nervous system
 In the fields of opto- and chemogenetics, scientists are always on the hunt for new actuators that are non-invasive and can rapidly and reversibly stimulate neurons. Recent studies have designed multi-component actuators that are sensitive to radio waves or allow for magnetogenetic control, but these actuators were not optimally designed for neuronal systems. To develop a new single-component tool for magnetogenetic control of neurons, the Guler Lab engineered an actuator in which two subunits of the paramagnetic ferritin protein were tethered to the C-terminus of TRPV4 (a pressure-sensitive channel). The Guler lab optimized the actuator’s subcellular localization using trafficking signals and named it Magneto 2.0.
In the fields of opto- and chemogenetics, scientists are always on the hunt for new actuators that are non-invasive and can rapidly and reversibly stimulate neurons. Recent studies have designed multi-component actuators that are sensitive to radio waves or allow for magnetogenetic control, but these actuators were not optimally designed for neuronal systems. To develop a new single-component tool for magnetogenetic control of neurons, the Guler Lab engineered an actuator in which two subunits of the paramagnetic ferritin protein were tethered to the C-terminus of TRPV4 (a pressure-sensitive channel). The Guler lab optimized the actuator’s subcellular localization using trafficking signals and named it Magneto 2.0.
Using Magneto2.0-p2A-mCherry constructs, the lab verified that Magneto2.0 was magnetically sensitive and could manipulate cell activity in vitro. The Guler lab went on to test Magneto2.0 in the mammalian brain after delivering it via AAV injection, finding that it could increase the activity of neurons from brain slice preparations. Magneto2.0 was also tested in vivo by expression in zebrafish and mice. These in vivo studies revealed that Magneto 2.0 allows for magnetic remote control of neuronal activation. Find Magneto 2.0 plasmids by following the links below.
- Wheeler M, et al. Nat. Neurosci 2016. PubMed PMID: 26950006
| Plasmid ID | Plasmid Name | Plasmid Type |
| 74309 | pcDNA3.0-TRPV4-p2A-ferritin-p2A-mCherry | Mammalian Expression |
| 74308 | pcDNA3.0-Magneto2.0-p2A-mCherry | Mammalian Expression |
| 74334 | pCR8-Magneto2.0-p2A-mCherry | Gateway |
| 74333 | pCR8-Magneto2.0 | Gateway |
| 74307 | pAAV-CMV-DIO-Magneto2.0-sNRPpA | AAV |
| 74306 | pAAV-CMV-DIO-TRPV4-p2A-ferritin-sNRPpA | AAV |
| 74302 | pDestTol2CG2-Neurog1-Magneto2.0-p2A-mCherry-pA | Zebrafish Expression |
Human kinase domain constructs for automated bacterial expression
 The misregulation of human kinases has been linked to several diseases, especially cancer. A great deal of chemical biology and drug discovery is focused on developing selective kinase inhibitors to probe these signaling pathways or develop potential therapeutics, but expressing and purifying human kinases remains challenging. Small molecule kinase inhibitors usually target soluble kinase domains, but these domains often do not express well in E. coli without their partner regulatory domains.
The misregulation of human kinases has been linked to several diseases, especially cancer. A great deal of chemical biology and drug discovery is focused on developing selective kinase inhibitors to probe these signaling pathways or develop potential therapeutics, but expressing and purifying human kinases remains challenging. Small molecule kinase inhibitors usually target soluble kinase domains, but these domains often do not express well in E. coli without their partner regulatory domains.
The Chodera Lab recently compiled the Human Kinase Domain Constructs Kit, a library of human kinase domain constructs developed through a large-scale expression screen that can be used to generate His-tagged human kinase constructs that express well in a simple bacterial expression system (Parton et. al. 2016). The key to obtaining high expression of the catalytic domains of each kinase is to express them with an appropriate phosphatase, a technique originally pioneered by Markus Seeliger for the high-yield expression of Src and Abl (Seeliger et. al. 2005). In this kit, Lambda phosphatase (Plasmid 79748) enhances bacterial expression of Serine/Threonine kinases while YopH (residues 164-468, Plasmid 79749) enhances expression of Tyrosine kinases. All sequences and expression data were kindly provided online by the Chodera Lab and the plasmids are now available through Addgene. All kinases in this library have crystallographic structures available in the PDB.
- Parton DL, Hanson SM, Rodríguez-Laureano L, Albanese SK, Gradia S, Jeans C, Seeliger MA, Levinson NM, Chodera JD. bioRxiv preprint. 2016. Full Text.
- Seeliger MA, Young M, Henderso MN, Pellicena P, King DS, Falick AM, and Kuriyan J. Protein Sci. 2005. PubMed PMID: 16260764

Topics: Addgene News





Leave a Comment