


This post was contributed by Jesse S. Boehm, the Associate Director of the Cancer Program at the Broad Institute of Harvard and MIT.
The notion of cancer precision medicine seems so simple! Take a patient’s tumor sample, use cutting edge genomic technologies to map the mutations that are present, and use prior knowledge (data connecting each genotype with vulnerabilities) to design a therapeutic strategy that works.
But, those darn cancers have revealed many tricks up their sleeves and most patients still don’t benefit from this approach. One central bottleneck is that most recurrently mutated cancer genes are rare and most of the individual variants found in tumors are exceedingly rare. As a result, how most of these “variants of unknown significance” (sometimes called “VUS”) function is unknown. How can we make a decision for each patient if the majority of information on each cancer clinical sequencing report includes rare variants that haven’t been characterized?
Some have developed exciting computational algorithms, such as SIFT or PolyPhen to approach this challenge, using attributes of cancer variants such as evolutionary conservation, or predicted deleteriousness of the amino acid change based on maps of protein structure. Sometimes this provides a great deal of clarity. But, often these methods don’t agree. Confusing indeed! We've developed a new plasmid toolkit, the Broad Institute Target Accelerator Plasmid Collection, that we hope researchers can use to characterize these variants in a high-throughput manner and therefore push us closer to making cancer precision medicine a reality.
Challenges for cancer variant characterization
For many years, we’ve hoped that large scale laboratory-based functional experiments that classify which variants are likely gain-of-function, loss-of-function or neutral, as compared to wild type, would be extremely helpful as a complementary approach. Also, in principle, these experiments could find correlations between variants in a gene of poorly classified function and a well established cellular signaling gene, thus providing a starting point for downstream validation experiments.
But, there have been two huge challenges preventing us from testing this hypothesis:
- There was no large scale, broadly available collection of mutant cDNA clones corresponding to cancer variants found in genome sequencing studies (as well as reference cDNA clones mapping to well-annotated cellular pathways).
- It wasn’t clear whether gene-agnostic bioassays could accurately test for molecular signatures of variant function and cancer phenotypes.
Broad Institute Target Accelerator Plasmid Collections
To overcome this first challenge, we’ve developed the Broad Institute Target Accelerator Plasmid Collections, now widely available to the scientific community through Addgene. These include 634 sequence confirmed mutant cDNA clones corresponding to cancer variants derived from analysis of lung adenocarcinoma genomes (Lung Cancer Mutant Collection) or “pan-cancer” analyses across many cancer types (Pan Cancer Mutant Collection). In addition, we worked with many collaborators to aggregate a sequence-confirmed series of clones corresponding to known cellular signaling pathways as part of our Genetic Pathway Reference Set. These Gateway-compatible entry clones are the product of many years of work and we’re proud to have worked closely with our Addgene colleagues to make these available to the scientific community.
Functional assays for cancer variants
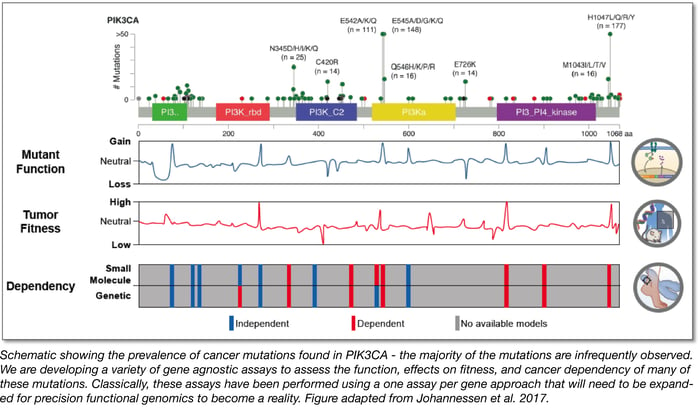
To overcome the second major challenge, we developed assays to test large numbers of cancer variants across a large number of genes as systematically as possible. In developing these assays, we figured that they would need to be generalizable and capable of measuring a variety of molecular signatures or cancer phenotypes. These assays were designed to complement the one assay per gene approach that has been the historical standard.
Specifically, in our recent publications, we used the cancer plasmid collections listed above to demonstrate how gene expression profiling, systematic tumorigenesis assays, drug epistasis assays, and morphological profiling can be used as generalizable strategies to map the function of rare cancer variants and genes:
- High-throughput Phenotyping of Lung Cancer Somatic Mutations. Berger AH, Brooks AN, Wu X, et al. Cancer Cell, 2016. PubMed PMID: 27478040.
- The lung cancer mutant collection is described in this paper.
- Systematic Functional Interrogation of Rare Cancer Variants Identifies Oncogenic Alleles. Kim E, Ilic N, Shrestha Y. et al. Cancer Discovery 2016. PubMed PMID: 27147599.
- The pan cancer mutant collection and the genetic pathway reference collections are described in this paper
- Systematic morphological profiling of human gene and allele function via Cell Painting. Rohban MH, Elife, 2017. PubMed PMID: 28315521. PubMed Central PMCID: PMC5386591.
- The genetic pathway reference collection is described in this publication
We also wrote an extensive review on the exciting new field we call “precision functional genomics.” The goal of this field is to functionally map the impact of cancer variants at scale. Check it out:
- Progress towards precision functional genomics in cancer. Johannessen, CM and Boehm JS. Curr. Opin Syst Biol. 2017. http://dx.doi.org/10.1016/j.coisb.2017.02.002.
We hope that by releasing these new and exciting plasmid collections (together with the data generated from these alleles in the above publications), it will be possible for many others to perform additional functional assays for these mutations and map other cellular functions and phenotypes.
Since time is of the essence in clinical settings, looking ahead, we’re starting to build saturating libraries of all possible mutations in many major cancer genes. These should be used to create ever more complete “look-up-tables” of mutant function that could enable future clinicians to improve real time decisions when rare variants are discovered in clinical sequencing studies. At the Broad, my colleague Cory Johannessen and his team have recently demonstrated the power of this approach:
- Phenotypic Characterization of a Comprehensive Set of MAPK1/ERK2 Missense Mutants. Brenan L et al. Cell Reports, 2016. PubMed PMID: 27760319. PubMed Central PMCID: PMC5120861.
While there is much work to do, these early pilot studies have demonstrated technical feasibility and proof-of-concept. If such approaches are proven to improve future clinical decisions, we can work together to realize the potential of precision functional genomics and propel the future of cancer precision medicine for the next-generation of cancer patients.
We hope you find these new cancer variant plasmid collections useful for your studies.
Many thanks to our guest blogger, Jess S. Boehm.
 Jesse S. Boehm is the Associate Director of the Cancer Program at the Broad Institute of Harvard and MIT and is focused on making precision functional genomics a reality for future cancer patients. You can follow him on Twitter @boehmjesse.
Jesse S. Boehm is the Associate Director of the Cancer Program at the Broad Institute of Harvard and MIT and is focused on making precision functional genomics a reality for future cancer patients. You can follow him on Twitter @boehmjesse.
Additional Resources on the Addgene Blog
- Learn About our Cancer Colleciton Pages
- Use the ClonTracer Library for Lineage Tracing
- Learn About Cancer Immunotherapies
Resources on Addgene.org
- Fine More Kits at Addgene
- Browse Plasmids from the Broad Institute

Topics: Cancer, Other Plasmid Tools, Plasmids



Leave a Comment